Este texto não substitui o publicado no Diário Oficial da União

Secretaria de Atenção à Saúde
PORTARIA Nº 55, DE 29 DE JANEIRO DE 2010
O Secretário de Atenção à Saúde, no uso de suas atribuições,
Considerando a necessidade de se melhorar os parâmetros sobre a Doença Falciforme no Brasil e de atualizar as diretrizes nacionais para diagnóstico, tratamento e acompanhamento dos indivíduos com esta doença;
Considerando que os Protocolos Clínicos e Diretrizes Terapêuticas (PCDT) são resultado de consenso técnico-científico e são formulados dentro de rigorosos parâmetros de qualidade, precisão de indicação e posologia;
Considerando as sugestões dadas à Consulta Pública SAS/MS nº 3, de 10 de novembro de 2009;
Considerando a Portaria SAS/MS nº 375, de 10 de novembro de 2009, que aprova o roteiro a ser utilizado na elaboração de PCDT, no âmbito da Secretaria de Atenção à Saúde - SAS; e
Considerando a avaliação do Departamento de Atenção Especializada - Secretaria de Atenção à Saúde, resolve:
Art. 1º Aprovar, na forma do Anexo desta Portaria, o PROTOCOLO CLÍNICO E DIRETRIZES TERAPÊUTICAS -DOENÇA FALCIFORME.
§ 1º O Protocolo objeto deste Artigo, que contêm o conceito geral da Doença Falciforme, critérios de diagnóstico, critérios de inclusão e de exclusão, tratamento e mecanismos de regulação, controle e avaliação, é de caráter nacional e deve ser utilizado pelas Secretarias de Saúde dos Estados e dos Municípios na regulação do acesso assistencial, autorização, registro e ressarcimento dos procedimentos correspondentes;
§ 2º É obrigatória a observância desse Protocolo para fins de dispensação do medicamento nele previsto;
§ 3º É obrigatória a cientificação do paciente, ou de seu responsável legal, dos potenciais riscos e efeitos colaterais relacionados ao uso do medicamento preconizado para o tratamento da Doença Falciforme, o que deverá ser formalizado por meio da assinatura do respectivo Termo de Esclarecimento e Responsabilidade, conforme o modelo integrante do Protocolo; e
§ 4º Os gestores estaduais e municipais do SUS, conforme a sua competência e pactuações, deverão estruturar a rede assistencial, definir os serviços referenciais e estabelecer os fluxos para o atendimento dos indivíduos com a doença em todas as etapas descritas no Anexo desta Portaria.
Art. 2º Esta Portaria entra em vigor na data de sua publicação.
Art. 3º Fica revogada a Portaria SAS/MS nº 872, de 06 de novembro de 2002, publicada no Diário Oficial da União, de 8 de novembro de 2002, seção 1, página 169.
ALBERTO BELTRAME
ANEXO
PROTOCOLO CLÍNICO E DIRETRIZES TERAPÊUTICAS
DOENÇA FALCIFORME
1. METODOLOGIA DE BUSCA DE LITERATURA
Para a análise de eficácia do tratamento específico para doença falciforme atualmente registrado na ANVISA e, portanto, disponível para utilização e comercialização no Brasil, foram realizadas as buscas nas bases descritas abaixo com a data limite de 30/08/2009.
Na base Medline/Pubmed:
- "hydroxyurea" [Substance Name] AND "hemoglobin, sickle" [Mesh]
- limitada a: "humans, meta-analysis, randomized controlled trial, review, journal article".
Na base Scielo:
-"hidroxiuréia" e "doença falciforme"
- limitada a: artigo original
2. INTRODUÇÃO
A doença falciforme (DF) é uma condição genética autossômica recessiva decorrente de defeitos na estrutura da hemoglobina (Hb) associada ou não a defeitos na sua síntese. (1,2) As hemoglobinopatias decorrentes dos defeitos na estrutura da Hb são mais frequentes em povos africanos, enquanto as talassemias decorrentesde defeitos na síntese da Hb, em povos do Mediterrâneo, da Ásia e da China.(1,2) Apesar dessa predileção étnica, a DF está presente em todos os continentes como consequência das migrações populacionais.(1-5) No Brasil, que reconhecidamente apresenta uma das populações de maior heterogeneidade genética do mundo (6), a maior prevalência da doença ocorre nas Regiões Norte e Nordeste.(7)
As pessoas com DF obrigatoriamente herdam uma mutação materna e outra paterna. As mutações herdadas podem estar em estado homozigótico (SS), único genótipo que pode ser denominado "anemia" falciforme (4,5), ou heterozigótico composto, ou seja, a doença é causada pela herança de Hb S em combinação com outro defeito (estrutural ou de síntese) na hemoglobina (SC, SD, SE, S beta-talassemia, S alfa-talassemia ou S mut rara). A maioria dos genitores de crianças com DF são heterozigotos simples, ou seja, apresentam um gene da Hb A (normal) associada com a hemoglobina variante.(1-5) Não é incomum a identificação de um dos pais como afetado pela DF durante a investigação familiar suscitada pelo nascimento de um filho diagnosticado através triagem neonatal ("teste do pezinho") para a doença.(8-10)
A heterogeneidade mutacional e outras características genéticas do indivíduo associadas a fatores ambientais e sociais são responsáveis por um amplo espectro de manifestações e complicações clínicas da DF (1,4,8) fato relevante que deve ser levado em consideração durante o complexo aconselhamento genético(10), bem como durante o tratamento e o acompanhamento clínico dos pacientes e de suas famílias.
O reconhecimento de que a DF é uma doença prevalente no Brasil (6,7) foi determinante na instituição da Política Nacional de Atenção à Doença Falciforme (PNADF) do Ministério da Saúde (MS).(11) Estima-se que 4% população brasileira tenha o traço falciforme (heterozigose simples) e que 25.000 a 50.000 pessoas tenham a doença em estado homozigótico (SS -anemia falciforme) ou na condição de heterozigotos compostos (SC; SE; SD; S/beta Talassemia - doença falciforme).(7)
A Hb S, na forma desoxigenada, perde sua complexa estrutura quaternária e adquire uma estrutura primária (polimerização hemoglobínica). A partir da sua polimerização, a HbS torna-se insolúvel, alterando a forma eritrocitária (que normalmente é um disco bicôncavo), para uma estrutura que lembra uma foice: fenômeno da eritrofalciformação. Os eritrócitos falciformados são fagocitados prematuramente pelo sistema monocítico-macrofágico, ocasionando anemia hemolítica crônica, geralmente, de importante magnitude.(12) A hemólise, por uma via metabólica complexa, compromete o metabolismo do óxido nítrico o que ocasiona vasculopatia proliferativa. Há alterações endotelias que geram um estado inflamatório crônico. O endotélio lesado expõe o fator tecidual, que desencadeia a cascata da coagulação e libera multímeros de von Willebrand. Ainda, na DF há expressão anômala de moléculas de adesão, havendo uma maior interação entre os elementos celulares sanguíneos e o endotélio vascular. (13-15)
Portanto, os pilares fisiopatogênicos da vaso-oclusão na DF são o fenômeno da eritrofalciformação, a maior interação entre células endoteliais, leucócitos e plaquetas, a vasculopatia proliferativa, o estado inflamatório crônico e a hipercoagulabilidade.(12-15)
A PNADF tem por objetivo diminuir a alta taxa de morbimortalidade da DF (11),que é caracterizada por uma anemia hemolítica crônica e por eventos agudos e potencialmente letais: crises vaso-oclusivas e síndrome torácica.(12) Por isso, o diagnóstico precoce através da triagem neonatal, o uso de imunobiológicos especiais e de antibioticoprofilaxia são ações fundamentais para a saúde das pessoas com a doença.(16)
A identificação dos pacientes antes do início sintomatológico visa diminuir os episódios vaso-oclusivos, também denominados de crises falcêmicas.(4,16) Nessas crises, pode haver dor intensa, lesões isquêmicas teciduais e danos em todos os órgãos e sistemas (cérebro, coração, fígado, rins, pele, olhos, esqueleto e pulmões) (17-20); sendo que a maioria dos desfechos fatais é precedida de episódios agudos, como, por exemplo, a síndrome torácica aguda.(3,12,21-33)
Em 1994, um estudo norte-americano multicêntrico denominado de Cooperative Study of Sickle Cell Disease (CSSCD) (23) observou que a sobrevida mediana dos homens com DF era de 42 anos e a das mulheres, de 48 anos; também constatou que as pessoas com a DF com valores de Hb fetal (Hb F) superiores a 8% sobreviviam mais do que aquelas com um valor abaixo desse ponto de corte.(24)
A hidroxiuréia (HU) apresenta um efeito citotóxico que ao inibir a enzima ribonucleotídeo redutase produz vários efeitos benéficos nos pacientes com DF, tais como: aumento da produção de HbF, aumento da hidratação do glóbulo vermelho, aumento da taxa hemoglobínica, maior produção de óxido nítrico e diminuição da expressão de moléculas de adesão. Até o momento, ela é considerada a terapia farmacológica de maior sucesso para a DF.(25-33)
Durante nove anos de observação, Steinberg e col. concluíram que a HU deve ser usada indefinidamente pelos pacientes de que dela necessitam(22), além disso, as chances do fármaco produzir efeitos indesejáveis são muito menores que as chances dessas pessoas apresentarem desfechos fatais em decorrência da doença.(21-34) O uso da HU diminuiu em 40% o risco de óbito pela DF(17,18), diminui de 4,5 para 2,5 o número anual de episódios álgicos agudos em adultos, reduz em cerca de 50% as necessidades transfusionais e o número de episódios de síndrome torácica aguda.(22,25)
A prescrição de HU para a população pediátrica tem demonstrado claros benefícios e parece ser bem tolerada(28-33), podendo prevenir nessa faixa etária tanto o infarto esplênico quanto as manifestações neurológicas (convulsões, paralisias, distúrbios da fala, cegueira transitória e alterações da consciência).(16,28-33)
As principais desvantagens do uso da HU são a necessidade de monitoramento frequente dos efeitos citotóxicos do fármaco, além disso, o potencial carcinogênico e teratogênico do fármaco tem um peso maior na hora de prescrição para crianças.(16,20,34,35) Outra desvantagem para a população infantil é a ausência da forma farmacêutica líquida da HU.(30)
A complexidade da DF e dos cuidados necessários durante o tratamento com HU suscitou a normatização da dispensação desse fármaco em centros de referência no tratamento da DF e a criação desse protocolo clínico e diretrizes terapêuticas desde 2002.(36)
3. CLASSIFICAÇÃO ESTATÍSTICA INTERNACIONAL DE DOENÇAS E PROBLEMAS RELACIONADOS À SAÚDE (CID-10)
- D56.1 Talassemia Beta
-D56.8 Outras talassemias
- D 57.0 Anemia falciforme com crise
- D 57.1 Anemia falciforme sem crise
- D 57.2 Transtornos falciformes heterozigóticos duplos
4. DIAGNÓSTICO
Tanto a eletroforese por focalização isoelétrica (IEF) quanto a cromatografia líquida de alta resolução (HPLC) podem ser utilizadas no diagnóstico para DF. Se HPLC tiver sido o método escolhido pelo programa de triagem neonatal da DF, os casos alterados nessa metodologia deverão ter sido confirmados por IEF e ser igualmente reportados.(37)
5. CRITÉRIOS DE INCLUSÃO
5.1. PREENCHER TODOS OS CRITÉRIOS ABAIXO RELACIONADOS:
- Idade superior a 3 anos;
- Condições de comparecer às consultas e de realizar exames laboratoriais periódicos;
- Teste de gravidez (beta-HCG sérico) negativo para mulheres em idade reprodutiva;
- Mulheres em idade reprodutiva devem se comprometer a usar método anticoncepcional com eficácia confirmada durante a terapia com HU.
5.2. PREENCHER PELO MENOS UM DOS SEGUINTES CRITÉRIOS NOS ÚLTIMOS 12 MESES:
- Três ou mais episódios álgicos agudos com necessidade de atendimento médico hospitalar ou comprovada incapacidade produtiva (escola/trabalho);
- Mais de um evento de síndrome torácica aguda (STA) definida como a presença de infiltrado pulmonar recente, não atelectásico, envolvendo pelo menos um segmento pulmonar completo, acompanhado de dor torácica, temperatura superior a 38,5 ºC, taquipnéia, sibilos, ou tosse em paciente com DF(3); ou um episódio de STA que necessitou de O2 ou transfusão sanguínea ou um episódio de STA que necessitou de internação em unidade de tratamento intensivo;
- Hipoxemia crônica: saturação de oxigênio persistentemente menor que 94% medida em duas visitas clínicas consecutivas, fora de um evento agudo e afastada a possibilidade de obstrução adenoidal/amigdaliana em crianças;
- Outras situações em que haja comprovação de lesão crônica de órgão (priapismo, necrose óssea, retinopatia proliferativa, entre outras);
- Concentração de Hb < 7g/dL (média de 3 valores fora de evento agudo);
- Concentração de Hb F < 8% após 2 anos de idade;
- Leucocitose > 20.000/mm3 (média de 3 valores medianos fora de evento agudo);
- Desidrogenase lática (DHL): 2 vezes acima do valor de referência para a idade;
- Alterações no ecodoppler transcraniano (>200cm/s com impossibilidade de regime transfusional crônico).
6. CRITÉRIOS DE EXCLUSÃO
- Hipersensibilidade à HU;
- Não apresentar níveis aceitáveis para o início do tratamento (Tabela 1), ou seja, contagem de neutrófilos inferior a 2.500mm3, de plaquetas inferior a 95.000mm3, de reticulócitos inferior a 95.000mm3 e hemoglobina menor que 4,5g/dL.
- Gestação: o uso de hidroxiuréia deve ser descontinuado três a seis meses antes da gestação devido aos possíveis efeitos teratogênicos do fármaco;(38)
- Aleitamento materno: Como a HU é excretada no leite humano, cabe decidir ou pela interrupção do aleitamento ou pela suspensão do uso do fármaco, levando-se em consideração a importância do tratamento para a mãe;
- Sorologia positiva para HIV: o uso concomitante de HU e anti-retrovirais aumenta o risco neuropatia periférica, pancreatite e insuficiência hepática, por isso, essa associação de medicamentos está contraindicada.
7. CASOS ESPECIAIS
Devido aos possíveis efeitos teratogênicos e carcinogênicos da HU, a instituição terapêutica em crianças menores de 3 anos de idade deve ser criteriosamente analisada, levando-se em consideração o risco de morbi-mortalidade da doença. Pelo menos um dos seguintes fatores devem estar presentes:
- dactilite (antes do primeiro ano de vida);
- concentração de Hb menor que 7g/dL (média de 3 valores fora de evento agudo);
- contagem de leucócitos maior que 20.000/mm3 (média de 3 valores fora de evento agudo).
8. CENTRO DE REFERÊNCIA
Conforme já definido na Portaria GM/MS nº 822, de 06 de junho de 2001 (37), os Programas Estaduais de Triagem Neonatal em Fase II são responsáveis pela triagem neonatal da DF. Em consonância com o Programa Nacional de Atenção Integral às Pessoas com Doença Falciforme definido na Portaria nº 1018/GM de 1º de julho de 2005 (39) e a Política Nacional de Atenção às Pessoas com Doença Falciforme e outras Hemoglobinopatias pela Portaria nº 1.391/GM de 16 de agosto de 2005 (11) são responsáveis pela promoção da saúde das pessoas com esta doença.
Finalmente, a regulamentação do Sistema Único de Saúde publicada no Diário Oficial da União de 4 de setembro de 2009 (40) diz que os pacientes com DF terão primeiro acompanhamento multidisciplinar em triagem neonatal (AMTN) com médico pediatra, psicólogo e assistente social. Suas famílias deverão receber orientação sobre o diagnóstico, o tratamento e devem ser encaminhadas para aconselhamento genético. A continuidade do atendimento deverá seguir o protocolo clínico e as diretrizes terapêuticas para tratamento da DF em centros de referência.
9. TRATAMENTO
9.1. FÁRMACO
A Hidroxiuréia (HU) está disponível em cápsulas em gel sólido contendo 500mg do princípio ativo.
Manipulação da preparação líquida para crianças: Recomenda-se dissolver a cápsula de 500mg de HU em 10ml de água destilada, obtendo a concentração de 50mg/mL, o que facilita a administração da dose correta por kilograma de peso.(33) A estabilidade química e funcional do medicamento é mantida por aproximadamente 6 meses em temperatura ambiente.(41) Por tratar-se de um medicamento citotóxico, é altamente recomendável que sejam seguidas as boas práticas de manipulação de preparações magistrais e oficinais.(42)
9.2 ESQUEMA DE ADMINISTRAÇÃO
9.2.1. DOSE INICIAL: 15mg/Kg/dia, dose única. Para o cálculo da dose utiliza-se o peso real ou o ideal, aquele que for menor.
9.2.2. DOSE MÁXIMA TOLERADA (DMT): A DMT não deve ser superior 35mg/Kg/dia. (19, 25-31) Ela é definida como a maior dose capaz de promover a melhora mais proeminente no curso clinico e laboratorial da doença, sem a ocorrência de toxicidade hematológica conforme Tabela 1.(25)
9.2.3. CONDUTA DURANTE A OCORRÊNCIA DE TOXICIDADE: a HU deve ser descontinuada até a recuperação hematológica, renal, hepática ou gastrointestinal.(25) A dose de reinício da terapêutica é de 5mg/Kg menor que a dose que estava utilizando quando apresentou a intoxicação, seguindo os mesmos critérios de controle até a dose máxima tolerada para cada específico, que poderá ser de 20, 25 ou 35mg/Kg/dia. (22,25)
9.3. TEMPO DE TRATAMENTO (CRITÉRIOS DE INTER-RUPÇÃO)
O tratamento deve ser de pelo menos 2 anos e mantido por tempo indeterminado de acordo com a resposta clinica e laboratorial. É importante lembrar que cerca de 25% das pessoas não apresentam resposta satisfatória à HU e, portanto, nestes casos esse tratamento deve ser suspenso.
9.4. BENEFÍCIOS ESPERADOS:
- Abolição ou diminuição dos episódios de dor;
- Aumento da produção de HbF;
- Aumento, mesmo que discreto, da concentração total da Hb;
- Diminuição dos episódios de síndrome torácica aguda;
- Diminuição do número de hospitalizações;
- Diminuição do número de transfusões sangüíneas;
- Regressão ou estabilização de danos em órgãos ou tecidos;
- Melhora do bem-estar e da qualidade de vida e maior sobrevida.
10 MONITORIZAÇÃO
10.1. EXAMES BASAIS (ANTES DO INÍCIO DO TRATA M E N TO ) :
- Hemograma com contagem de plaquetas e reticulócitos para avaliar a inclusão e toxicidade da HU;
- Eletroforese de Hb com dosagem de HbF - para avaliar os efeitos benéficos do tratamento;
- Sorologias para hepatite B, hepatite C e HIV;
- Dosagem sérica de transaminases (AST, ALT) e creatinina;
- Dosagem de ácido úrico;
beta-HCG sérico.
10.2. MONITORIZAÇÃO LABORATORIAL
10.2.1 Hemograma completo e contagem de reticulócitos (ver Tabela 1):
- até dose de manutenção: a cada 2 semanas;
- após dose de manutenção: a cada 4 semanas.
10.2.2. Creatinina e transaminases (AST, ALT): considera-se toxicidade renal quando a dosagem de creatinina é superior a 50% ao valor basal e toxicidade hepática quando o valor de ALT é maior que duas vezes o limite superior.
- até dose de manutenção: a cada 4 semanas;
- após dose de manutenção: a cada 12 semanas.
10.2.3. Hemoglobina Fetal (ver Tabela 1):
- até dose de manutenção: a cada 8 semanas;
- após dose de manutenção: a cada 24 semanas.
Devido aos possíveis efeitos adversos do fármaco, a relação entre risco e o benefício deve ser cuidadosamente avaliada nos seguintes casos:
- Uricosúria: uso de HU pode aumentar os níveis séricos de ácido úrico. Em pessoas com níveis basais acima do limite normal estes valores devem ser monitorados mensalmente;
- Ácido fólico: o uso de HU produz macrocitose, dificultando o suspeita laboratorial de deficiência de ácido fólico. Desta forma, é recomendado o emprego profilático concomitante de 5mg/dia de ácido fólico, três vezes por semana. Cabe ressaltar que a deficiência do ácido fólico aumenta o risco de defeitos congênitos, especialmente de fechamento do tubo neural;
- Interações medicamentosas: não há estudos adequados sobre interação entre HU e outros medicamentos. Portanto, o uso concomitante de outros fármacos, principalmente os que também possam produzir depressão da medula óssea, deve ser cuidadosamente monitorizado;
- Sorologia positiva para HIV: a associação de HU com a antiretrovirais, a didanosina e a estavudina está contra-indicada;
- Sorologia positiva para hepatite B e C: as provas de função hepática devem ser monitoradas mensalmente durante o uso da HU; no caso de insuficiência hepática não há recomendação de ajuste da dose. O uso de hidroxiuréia pode diminuir os efeitos colaterais provocados por fármacos antivirais usados no tratamento das hepatites;(43)
- Insuficiência renal: embora poucos estudos tenham avaliado o uso de HU em pessoas com insuficiência renal, recomenda-se o ajuste de dose conforme o valor de depuração da creatinina: 1050mL/min administrar 50% da dose; <10mL/min administrar 20% da dose. É recomendável a avaliação em conjunto com o nefrologista. Pessoas em hemodiálise devem receber HU após o procedimento;
- Medidas antopométricas: o peso, a altura e o perímetro cefálico das crianças devem ser monitorados a cada 2 semanas durante os 2 primeiros meses de tratamento ou enquanto a dose estiver sendo ajustada; quando a criança estiver com sua dose de manutenção, este monitoramento deve ser feito a cada mês. De acordo com Tompson et AL (21), crianças com idades entre 9 e 17 meses devem ser realizar testes de desenvolvimento neuropsicomotor, pois dados não publicados de pesquisas em animais sugerem que a HU possa provocar um efeito deletério no crescimento e desenvolvimento cerebral.
Todos os eventos adversos (EA) relacionados ao uso de HU devem ser valorizados (44), pois podem contribuir para uma má aderência ao tratamento. O uso da HU pode ser mantido na vigência de EA leve, desde que haja acompanhamento regular de um especialista, porém a ocorrência de EA moderado ou grave exige suspensão do uso passível de reintrodução na dependência do dano causado e da vontade do usuário.
Neurológicos: letargia, cefaléia, tonturas, desorientação e raramente, alucinações;
Gastrintestinais: estomatite, anorexia, náuseas, vômitos, diarréia e constipação;
Dermatológicos: erupção maculopapular, eritema facial e periférico, alopecia, hiperpigmentação dos anexos (pele e das unhas), pele seca, ulceração da pele ou agravamento de úlcera já existente.Úlcera isquêmica é um possível efeito adverso do uso crônico de HU. Pessoas com história prévia ou atual de úlcera isquêmica, não há contra-indicação formal do uso de HU, entretanto, aparecimento de úlcera isquêmica sem história anterior desta complicação, a suspensão da HU deve ser considerada;(45)
Renais: elevação dos níveis séricos de uréia e creatinina;
Hepáticos: elevação das aminotransferases;
Reprodutivo: oligospermia, azoospermia; efeito teratogênico fetal;
Hematológicos: mielotoxicidade e hiperesplenismo em crianças;
Outros: edema, febre, calafrios, mal-estar, astenia.
Tabela 1. Ajuste da dose diária de hidroxiuréia*, de acordo com a contagem de neutrófilos, contagem de plaquetas, nível da hemoglobina e contagem de reticulócitos.
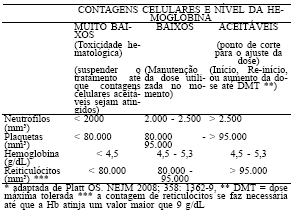
11. Regulação/controle/avaliação pelo gestor
Recomenda-se que as pessoas de qualquer idade com diagnóstico de DF sejam acompanhadas no Centro de Referência para Doença Falciforme, que facilita o tratamento em si, bem como o manejo das doses conforme necessário e o controle de efeitos ad-versos. O Centro de Referência que contar com farmacêutico poderá dispensar a HU diretamente para o paciente.
12. TERMO DE ESCLARECIMENTO E RESPONSABILIDADE
É obrigatória a cientificação do paciente, ou de seu responsável legal, dos potenciais riscos, benefícios e efeitos colaterais relacionados ao uso do medicamento preconizado neste Protocolo. O TER é obrigatório ao se prescrever medicamento do Componente Especializado da Assistência Farmacêutica.
13. REFERÊNCIAS BIBLIOGRÁFICAS
1. Weatherall DJ, Clegg JB. Inherited hemoglobin disorders: a increasing global health problem. Bulletin of the World Health Organization, 2001, 79 (8): 704-712.
2. Weatherall D, Akimyanju O, Fucharoen S, Olivieri N, Musgrove P. Inherited disorders of hemoglobin. In: Non-Communicable Diseases (Disease Control priorities in developing countries. Oxford Univ Press, New York, NY), 2nd Ed, pp 663-680. download at: http://files.dcp2.org/pdf/expressbooks/noncomm.pdf em 29 de julho de 2009.
3. Platt OS. The acute chest syndrome of sickle cell disease. (editorial) New England J. Med. 2000. 342: 1904-1907.
4. Zago MA, Pinto ACS. Fisiopatologia das doenças falciformes: da mutação genética à insuficência de múltiplos órgãos. Rev bras hematol hemoter, 2007; 29(3):207-214.
5. Schechter AN. Hemoglobin research and the origins of molecular medicine. Blood, 2008; 112 (10): 3927-3938. Download : www.bloodjournal.org em 31 de julho de 2009.
6. Parra FC, Amado RC, Lambertucci JR, Rocha J, Antunes CM, Pena SDJ..Color and genomic ancestry in Brazilians. Proc Natl Acad Sci U S A 2003; 100(1): 177-82.
7.Cançado RD, Jesus JA. A doença falciforme no Brasil. Rev bras hematol hemoter, 2007; 29(3):204-206.
8. de Paiva e Silva RB; Ramalho AS, Cassorla RMS. A anemia falciforme como problema de Saúde Pública no Brasil. Rev. Saúde Pública [online]. 1993;27 (1):54-58.
9. Silla LMR. Doença Falciforme: um grave e desconhecido problema de saúde pública no Brasil. Jornal de Pdiatria. 1999; 75 (3): 145-146.
10. Guedes C, Diniz D. Um caso de discriminação genética: o traço falciforme no Brasil PHYSIS: Rev. Saúde Coletiva. 2007; 17(3):501-520.
11. BRASIL. Ministério da Saúde. Portaria nº 1.391/GM de 16 de agosto de 2005 Disponível em: <http://dtr2001.saude. gov.br/sas/PORTARIAS/Port2005/GM/GM-1391htm>. Acesso em: 20 ago. 2009.
12. Gladwin MR, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008; 359; 21: 2254-2265.
13. Wood KC, Granger DN. Sickle cell disease: role of reactive oxygen and nitrogen metabolites. Clin Exp Pharmacol Physiol. 2007 ;34(9):926-32;
14. Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004; 11(2):129-51;
15. Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21(1):37-47.
16. Vichinsky E, Hurst D, Earles A, Keman K, Lubin B. Newborn screening for sickle cell disease: effect on mortality. Pediatrics, 1988; 81(6):749-755.
17. Strouse JJ, Lanzkron S, Beach MC Strouse JJ, Lanzkron S, Beach MC, Haywood C, Park H, Witkop C, Wilson RF, Bass. Hydroxyurea for sickle cell disease: a systematic review for efficacy and toxicity in children. Pediatrics 2008; 122:1332-1342.
18. Agência Nacional de Vigilância Sanitária (ANVISA). Manual de diagnóstico e tratamento de doenças falciformes. Brasília, DF: Ministério da Saúde; 2002.
19. Platt O, Brambilla DJ, Rosse WF et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994; 330:1639-1644.
20. Hankins JS, Ware RE, Rogers ZR et al. Long-term hydroxyurea therapy for infants with sickle cell anemia: the HUSOFT extension study. Blood 2005;106(7):2269-75.
21. Thompson BW, Miller ST, Roger ZR, Ree RC, Ware RE, Waclawiw MA, Iyer RV, Casella JF, Luchteman-Jones L, Rana S, Thornburg CD, Kalpatthi RV, Barredo JC, Brown RC, Sarnaik S, Howard TH, Luck L, Wang WC. The pediatric hydroxyurea phase III clinical trial (Baby HUG): Challenges of study design. Pediatr Blood Cancer 2009; DOI 10.1002/phc
22. Steinberg MH, Barton F, Castro O, Pegelow CH, Balas SK, Kutlar A, Orringer E, Bellevue R, Olivieri N, Wckman J, Varma M, Ramirez G, Adler B, Smith W, Carlos T, Atag K, de Castro L, Bigelow C, Sauntharajah Y, Telfer M, Vichinsky E, Claster S, Shurin S, Bridges K, Waclawiw M, Bonds D, Terrin M.. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA 2003; 289 (13):1645-1651.
23. Charache S, Terrin ML, Moore RD et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. NEngl J Med 1995;332(20):1317-22
24. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease - life expectancy and risk factors for early death. N Engl J Med. 1994 Jun 9;330(23):1639-44.
25. Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med. 2008. 358(13): 1362-1369.
26. Vicari P, Barretto de Mello A, and Figueiredo MS. Effects of hydroxyurea in a population of Brazilian patients with sickle cell anemia. Am J Hematol 2005 ;78(3):243-4.
27. Lanzkron S, Strouse JJ, Wilson R, Beach MC, Haywood C. Systematic Review: hydroxyrea for the treatment of adults with sickle cell disease. Ann Inter Med 2008; 148:939-955
28. Zimmerman SA, Schultz WH, Davis JS et al. Sustained long-term hematologic efficacy of hydroxyurea at maximum tolerated dose in children with sickle cell disease. Blood 2004;103(6):2039-45.
29. Zimmerman SA, Schultz WH, Burgett S et al. Hydroxyurea therapy lowers transcranial Doppler flow velocities in children with sickle cell anemia. Blood 2007, 110: 1043-1047.
30. Ferster A, Tahriri P, Vermylen C et al. Five years of experience with hydroxyurea in children and young adults with sickle cell disease. Blood 2001;97(11):3628-32.
31. de Montalembert M, Brousse V, Elie C et al. Long-term hydroxyurea treatment in children with sickle cell disease: tolerance and clinical outcomes. Haematologica 2006;91(1):125-8.
32. Hankins JS, Helton KJ, McCarville MB et al. Preservation of spleen and brain function in children with sickle cell anemia treated with hydroxyurea. Pediatr Blood Cancer 2007.
33. Gulbis B, Haberman D, Dufour D et al. Hydroxyurea for sickle cell disease in children and for prevention of cerebrovascular events: the Belgian experience. Blood 2005;105(7):2685-90.
34. Mueller BU. When should hydroxyrea be used for children with sickle cell disease? Pediatrics 2008; 122:1365-1366.
35. Friedrisch JR, Prá D, Maluf SW, Bittar CM, Mergener M, Pollo T, Kayser M, da Silva MA, Henriques JA, da Rocha Silla LM. DNA damage in blood leukocytes of individuals with sickle cell disease treated with hydroxyurea. Muta Res. 2008; 649:213-20.
36. BRASIL. Ministério da Saúde. Portaria GN/MS no 872 de 6 de novembro de 2002. Protocolo Clínico e Diretrizes Terapêuticas - Doença Falciforme - Hidroxiuréia. Disponível em: < http:// dtr2001. saude. gov. br/ sas/ PORTARIAS/ Port2002/ GM/ GM- 872htm>. Acesso em: 20 ago. 2009.
37. BRASIL. Ministério da Saúde. Portaria GM/MS nº 822/GM de 06 de junho de 2001. Disponível em: <http://dtr2001.saude. gov.br/sas/PORTARIAS/Port2001/GM/GM-822htm>. Acesso em: 20 ago. 2009.
38. Zanette AM. Gravidez e contracepção na doença falciforme. Rev. brasil. hematol. hemoter. 2007; 29 (3): 309-312.
39. BRASIL. Ministério da Saúde. Portaria nº 1.018/GM de 1º de Julho de 2005 Disponível em: <http://dtr2001.saude. gov.br/sas/PORTARIAS/Port2005/GM/GM-1018htm>. Acesso em: 20 ago. 2009.
40. BRASIL. Ministério da Saúde. Artigos 187,188, 322-331 da Portaria n nº 2048/GM de 3 de setembro de 2009. REGULAMENTAÇÃO DO SISTEMA ÚNICO DE SÁUDE Diário Oficial da União de 3 de setembro de 2009. . Disponível em:< http://www.in.g o v. b r / a u t e n t i c i d a d e.h t m l, pelo código 00012009090400158>
41 Heeney MM, Whorton MR, Howard TA, Johnson CA and Ware RE. Chemical and functional analysis of hydroxyurea oral solutions. J Pediatr Hematol Oncol 2004. 26(3): 179-84.
42. Brasil. Ministério da Saúde. Agência Nacional de Vigilância Sanitária - ANVISA. Resolução da diretoria colegiada (RDC) Nº 67 de 8 de outubro de 2007. Disponível em: < https://anvisa. gov. br/ legis/ resol/ 2007/ rdc/ 67_ 081007rdc. htm.
43. Hassan M, Hasan S, Castro O, Giday S, Banks A, Smoot D. HCV in sickle cell disease. J Natl Med Assoc 2003. 95(9): 872-874.
44. Bandeira FMGC, Peres JC, Carvalho EJ, Bezerra I, Araújo AS Mello MRB, Machado C. Hidroxiuréia em pacientes com síndromes falciformes acompanhados no Hospital Hemope, Recife, Brasil. Rev. Bras. Hematol. Hemoter. 2004. 26 (3): 189-194. [download: 2009-09-16], pp. 189-194.
45.Chaine B, Neonato MG, Girot R et al. Cutaneous adverse reactions to hydroxyurea in patients with sickle cell disease. Arch Dermatol 2001;137(4):467-70.
TERMO DE ESCLARECIMENTO E RESPONSABILIDADE
HIDROXIURÉIA
Eu, ________________________________________ (nome do(a) paciente), declaro ter sido informado(a) claramente sobre os benefícios, riscos, contra-indicações e principais efeitos adversos relacionados ao uso do medicamento hidroxiuréia, indicado para o tratamento da doença falciforme.
Os termos médicos foram explicados e todas as minhas dúvidas foram resolvidas pelo médico _____________________________________________________(nome do médico que prescreve).
Assim declaro que:
Fui claramente informado(a) de que o medicamento hidroxiuréia que passo a receber pode trazer as seguintes melhorias:
- Desaparecimento ou diminuição dos episódios de dor;
- Aumento da produção de Hb fetal;
- Aumento, mesmo que pequeno, da concentração total da Hb;
- Diminuição dos episódios de síndrome torácica aguda;
- Diminuição do número de hospitalizações;
- Diminuição do número de transfusões sangüíneas;
- Regressão ou estabilização de danos em órgãos ou tecidos;
- Melhora do bem-estar e da qualidade de vida e maior sobrevida.
Fui também claramente informado a respeito das seguintes contra-indicações, potenciais efeitos adversos e riscos:
- podem ocorrer os seguintes efeitos adversos: diminuição do número de glóbulos brancos (leucopenia e neutropenia), de glóbulos vermelhos (anemia) e do número de plaquetas, cansaço, dor de cabeça, tonturas, desorientação e alucinações; perda de apetite, náuseas, vômitos, diarréia, prisão de ventre e dor de estômago; elevação de enzimas hepáticas, hepatite medicamentosa, infiltrado pulmonar e fibrose pulmonar, erupções na pele, hiperpigmentação das unhas, queda de cabelos, câncer de pele; perda de função renal, elevação dos níveis sangüíneos de uréia, creatinina e ácido úrico; febre, calafrios, mal-estar.
- a hidroxiuréia atravessa a placenta e vai para circulação fetal e isso tem um potencial risco de causar defeitos congênitos no feto e até óbito fetal, por isso a gravidez deve ser evitada durante o tratamento com a hidroxiuréia; Reprodução: pode causar redução da capacidade reprodutiva de homens e mulheres;
- a hidroxiuréia é excretada através do leito materno e é totalmente contra-indicado seu uso em crianças sadias; existem duas opções a serem discutidas individualmente: suspensão do aleitamento materno ou suspensão do fármaco - levar em consideração os efeitos benéficos do aleitamento e do tratamento para a mãe.
Estou ciente de que a hidroxiuréia somente pode ser utilizado por mim, comprometendo-me a devolvê-lo caso não queira ou não possa utilizá-lo ou se o tratamento for interrompido. Sei também que continuarei ser atendido, inclusive em caso de eu desistir de usar o medicamento.
Também estou ciente de que o ácido fólico, medicamento complementar ao meu tratamento, pode, raramente, ser maléfico à minha função renal, além de provocar reação alérgica (febre e erupção cutânea).
Autorizo o Ministério da Saúde e as Secretarias de Saúde a
fazer uso de informações relativas ao meu tratamento, desde que
assegurado o anonimato.
| Local: Data: | ||
| Nome do paciente: | ||
| Cartão Nacional de Saúde: | ||
| Nome do responsável legal: | ||
| Documento de identificação do responsável legal: | ||
| Assinatura do paciente ou do responsável legal | ||
| Médico Responsável: | CRM: | UF: |
| Assinatura e carimbo do médico Data:____________________ |
||
Observação: Este Termo é obrigatório ao se solicitar o fornecimento de medicamento do Componente Especializado da Assistência Farmacêutica e deverá ser preenchido em duas vias, ficando uma arquivada na farmácia e a outra entregue ao usuário ou seu responsável legal.