Este texto não substitui o publicado no Diário Oficial da União

Secretaria de Atenção à Saúde
PORTARIA Nº 492, DE 23 DE SETEMBRO DE 2010
O Secretário de Atenção à Saúde, no uso de suas atribuições,
Considerando a necessidade de se estabelecer parâmetros sobre a epilepsia no Brasil e de diretrizes nacionais para diagnóstico, tratamento e acompanhamento dos indivíduos com esta doença;
Considerando que os Protocolos Clínicos e Diretrizes Terapêuticas (PCDT) são resultado de consenso técnico-científico e são formulados dentro de rigorosos parâmetros de qualidade, precisão de indicação e posologia;
Considerando as sugestões dadas à Consulta Pública SAS/MS no 23, de 10 de maio de 2010;
Considerando a Portaria SAS/MS nº 375, de 10 de novembro de 2009, que aprova o roteiro a ser utilizado na elaboração de PCDT, no âmbito da Secretaria de Atenção à Saúde - SAS; e
Considerando a avaliação do Departamento de Atenção Especializada - DAE/SAS, resolve:
Art. 1º Aprovar, na forma do Anexo desta Portaria, o PROTOCOLO CLÍNICO E DIRETRIZES TERAPÊUTICAS - EPILEPSIA.
§ 1º O Protocolo, objeto deste Artigo, que contêm o conceito geral da epilepsia, critérios de diagnóstico, critérios de inclusão e de exclusão, tratamento e mecanismos de regulação, controle e avaliação, é de caráter nacional e deve ser utilizado pelas Secretarias de Saúde dos Estados e dos Municípios na regulação do acesso assistencial, autorização, registro e ressarcimento dos procedimentos correspondentes.
§ 2º É obrigatória a observância desse Protocolo para fins de dispensação de medicamento nele previsto.
§ 3º É obrigatória a cientificação do paciente, ou de seu responsável legal, dos potenciais riscos e efeitos colaterais relacionados ao uso de medicamento preconizado para o tratamento da epilepsia, o que deverá ser formalizado por meio da assinatura do respectivo Termo de Esclarecimento e Responsabilidade, conforme o modelo integrante do Protocolo.
§ 4º Os gestores estaduais e municipais do SUS, conforme a sua competência e pactuações, deverão estruturar a rede assistencial, definir os serviços referenciais e estabelecer os fluxos para o atendimento dos indivíduos com a doença em todas as etapas descritas no Anexo desta Portaria.
Art. 2º Esta Portaria entra em vigor na data de sua publicação.
Art. 3º Fica revogada a Portaria SAS/MS nº 864, de 04 de novembro de 2002, publicada no Diário Oficial da União nº 217, de 8 de novembro de 2002, Seção 1, página 163.
ALBERTO BELTRAME
ANEXO
PROTOCOLO CLÍNICO E DIRETRIZES TERAPÊUTICAS
EPILEPSIA
1. METODOLOGIA DE BUSCA DA LITERATURA
Foram realizadas buscas no Medline/Pubmed com as palavras-chave "epilepsy" [Mesh] AND "drug therapy" [Mesh] AND"anticonvulsants" [Mesh]. Todos os estudos encontrados metanálises e ensaios clínicos randomizados, controlados , publicados até 31/01/2010, foram avaliados.
Quando usados limites de metanálises, ensaios clínicos randomizados, em língua inglesa, publicados a partir de 2004, foram encontrados 236 artigos. Apenas os que referiam medicamentos disponíveis no Brasil foram considerados. Em análise mais acurada, foram excluídos outros procedimentos, estudos primariamente de outras doenças neurológicas, outras metodologias (estudos abertos, comparações entre tratamentos precoces e tardios) e estudos com outros desfechos principais que não controle de crises (sono, reprodução, função cardíaca, refratariedade ao tratamento, cognição, gestação, funções executivas, alterações do sistema imunológico, emoções, qualidade de vida, osteoporose, descargas epileptiformes interictais, retirada de medicamentos), chegando-se a um total de 67 artigos elegíveis para referências bibliográficas.
Na base de dados Cochrane, em acesso realizado em 04/03/2010, utilizando-se a estratégia "epilepsy" AND "pharmacological treatment". foram identificadas 52 revisões sistemáticas que, avaliadas individualmente, resultaram em 17 revisões consideradas relevantes e incluídas na elaboração do protocolo.
Foram também utilizados livros-textos de Neurologia e artigos não indexados.
2. INTRODUÇÃO
Epilepsia é uma doença cerebral crônica causada por diversas etiologias e caracterizada pela recorrência de crises epilépticas não provocadas(1). Esta condição tem consequências neurobiológicas, cognitivas, psicológicas e sociais e prejudica diretamente a qualidade de vida do indivíduo afetado(2).
Estima-se que a prevalência mundial de epilepsia ativa esteja em torno de 0,5%-1,0% da população(3) e que cerca de 30% dos pacientes sejam refratários, ou seja, continuam a ter crises, sem remissão, apesar de tratamento adequado com medicamentos anticonvulsivantes(4). A incidência estimada na população ocidental é de 1 caso para cada 2.000 pessoas por ano. A incidência de epilepsia é maior no primeiro ano de vida e volta a aumentar após os 60 anos de idade. A probabilidade geral de ser afetado por epilepsia ao longo da vida é de cerca de 3%(5). No Brasil, Marino e colaboradores6 e Fernandes e colaboradores7 encontraram prevalências de 11,9:1.000 na Grande São Paulo e de 16,5:1.000 para epilepsia ativa em Porto Alegre.
De forma prática, as epilepsias podem ser classificadas segundo dois grandes eixos: topográfico e etiológico. No eixo topográfico, as epilepsias são separadas em generalizadas e focais. As generalizadas manifestam-se por crises epilépticas cujo início envolve ambos os hemisférios simultaneamente. Em geral, são geneticamente determinadas e acompanhadas de alteração da consciência; quando presentes, as manifestações motoras são sempre bilaterais. Crises de ausência, crises mioclônicas e crises tônico-clônicas generalizadas (TCG) são seus principais exemplos (8).
Nas epilepsias focais, as crises epilépticas iniciam de forma localizada numa área específica do cérebro, e suas manifestações clínicas dependem do local de início e da velocidade de propagação da descarga epileptogênica. As crises dividem-se em focais simples (sem comprometimento da consciência) e focais complexas (com comprometimento ao menos parcial da consciência durante o episódio). Por fim, uma crise focal, seja simples ou complexa, quando propagada para todo o córtex cerebral, pode terminar numa crise TCG, sendo então denominada crise focal secundariamente generalizada9.
No eixo etiológico, as epilepsias são divididas em idiopáticas (sem lesão estrutural subjacente), sintomáticas (com lesão) ou criptogênicas (presumivelmente sintomáticas, mas sem uma lesão aos exames de imagem disponíveis no momento) (10). As causas lesionais mais frequentes das epilepsias focais sintomáticas são esclerose temporal mesial, neoplasias cerebrais primárias, anomalias vasculares e malformações do desenvolvimento corticocerebral (11).
Na infância, situações relativamente benignas, como epilepsia rolândica benigna da infância e convulsões febris simples, podem ocorrer. Encefalopatias epilépticas, tais como as síndromes de West e de Lennox-Gastaut, estão comumente associadas a alguma doença de base (são, portanto, sintomáticas na sua maioria) e geralmente apresentam mau prognóstico tanto do ponto de vista do controle medicamentoso de crises como no tocante ao desenvolvimento neuropsicomotor(12).
A epilepsia rolândica benigna da infância geralmente tem início na pré-adolescência (7 a 10 anos de idade), predomina em meninos (numa proporção de 1,5:1) e apresenta alta prevalência (15% das epilepsias da infância). As crises caracterizam-se por sintomas motores ou sensoriais faciais unilaterais, manifestações orofaríngeas, paralisia da fala e hipersalivação. É uma epilepsia geneticamente determinada, com evidências de ligação com o cromossoma 15q14. Sua herança é autossômica dominante, com penetrância idade-dependente. Apesar de clinicamente as crianças terem aspecto muito próximo do normal, o eletroencefalograma mostra-se desproporcional e gravemente alterado, com atividade epileptogênica localizada em uma ou em ambas as regiões centrais, sobretudo durante o sono. O prognóstico é excelente: o risco de desenvolver crises generalizadas infrequentes na idade adulta é inferior a 2%. A remissão das crises ocorre 2-4 anos após o início do quadro e sempre antes dos 16 anos de idade. A maioria dos pacientes apresenta menos de 10 crises ao longo do tratamento (13).
Convulsões febris devem-se a uma suscetibilidade aumentada a crises epilépticas, são idade-dependente (6 meses - 5 anos) e geneticamente determinadas. As crises são precipitadas por febre, sem evidência de infecção do sistema nervoso central (SNC) ou outra causa. Há uma leve predominância do sexo masculino (60%). A prevalência é de cerca de 3% das crianças. As crises duram menos de 15 minutos, são generalizadas e não se acompanham de deficits neurológicos. Não há necessidade de medicamentos anticonvulsivantes preventivos. Cerca de 3% das crianças que tiveram crises febris desenvolvem algum tipo de epilepsia na idade adulta. Em geral, o prognóstico é bom, com desenvolvimentos cognitivo e comportamental adequados (14).
A síndrome de West é uma encefalopatia epiléptica relacionada especificamente a crianças com menos de 1 ano de idade, resultante de múltiplas causas. Ela é caracterizada por um tipo específico de crise epiléptica, denominada "espasmos epilépticos", e anormalidades grosseiras ao eletrocardiograma (o chamado padrão hipsarrítmico ou hipsarritmia). O desenvolvimento psicomotor é invariavelmente prejudicado. Cerca de 60% das crianças desenvolvem outros tipos de crises, evoluindo para síndrome de Lennox-Gastaut (15).
Lennox-Gastaut é uma síndrome da infância caracterizada pela tríade: crises epilépticas polimorfas intratáveis (em geral, tônicas, atônicas ou de ausência atípica), anormalidades cognitivas e comportamentais e EEG com paroxismos de atividade rápida e descargas generalizadas de complexos onda aguda-onda lenta a 2,5 Hz. Manifesta-se antes dos 11 anos de idade, e as crises geralmente resultam em quedas. A exemplo da de West, esta síndrome também apresenta uma vasta lista de possíveis etiologias. O prognóstico é ruim, com 5% de mortalidade. Cerca de 80%-90% dos pacientes continuam a apresentar crises epilépticas na vida adulta(16).
3. CLASSIFICAÇÃO ESTATÍSTICA INTERNACIONAL DE DOENÇAS E PROBLEMAS RELACIONADOS À SAÚDE - CID 10
- G40.0 Epilepsia e síndromes epilépticas idiopáticas definidas
por sua localização (focal) (parcial) com crises de início focal;
- G40.1 Epilepsia e síndromes epilépticas sintomáticas definidas
por sua localização (focal) (parcial) com crises parciais simples;
- G40.2 Epilepsia e síndromes epilépticas sintomáticas definidas
por sua localização (focal) (parcial) com crises parciais complexas;
- G40.3 Epilepsia e síndromes epilépticas generalizadas idiopáticas;
- G40.4 Outras epilepsias e síndromes epilépticas generalizadas;
- G40.5 Síndromes epilépticas especiais;
- G40.6 Crise de grande mal, não especificada (com ou sem
pequeno mal);
- G40.7 Pequeno mal não especificado, sem crises de grande
mal; ou
- G40.8 Outras epilepsias.
4. DIAGNÓSTICO
4.1. CLÍNICO
Na maioria dos casos, o diagnóstico de uma crise epiléptica pode ser feito clinicamente através da obtenção de uma história detalhada e de um exame físico geral, com ênfase nas áreas neurológica e psiquiátrica. Muitas vezes, o auxílio de uma testemunha ocular é importante para que a crise seja descrita em detalhes. A existência de aura bem como as condições que possam ter precipitado a crise devem ser registradas. Idade de início, frequência de ocorrência e intervalos mais curtos e mais longos entre as crises devem ser caracterizados, muitas vezes com o auxílio de um diário de crises. A história deve cobrir a existência de eventos pré e perinatais, crises no período neonatal, crises febris, qualquer crise não provocada e história de epilepsia na família. Trauma craniano, infecção ou intoxicações prévias também devem ser investigados(9).
É fundamental um diagnóstico diferencial correto com outros distúrbios paroxísticos da consciência, como síncopes e crises não epilépticas psicogênicas.
4.2 COMPLEMENTAR
Os exames complementares devem ser orientados pelos achados da história e do exame físico. O principal exame é a eletroencefalografia (EEG), cujo papel é auxiliar o médico a estabelecer um diagnóstico acurado. EEG é capaz de responder a três importantes questões diagnósticas nos pacientes com suspeita de epilepsia: 1) o paciente tem epilepsia?, 2) onde está localizada a zona epileptogênica? e 3) o tratamento está sendo adequado?(17).
Exames de imagem (ressonância magnética (RM) do encéfalo e tomografia computadorizada (TC) de crânio) devem ser solicitados na suspeita de causas estruturais (lesões cerebrais, tais como tumores, malformações vasculares ou esclerose hipocampal), que estão presentes na maioria dos pacientes com epilepsia focal(18). O diagnóstico de uma causa estrutural subjacente tem implicações terapêuticas as quais podem embasar a indicação de tratamento cirúrgico da epilepsia, e prognósticas, definindo mais precocemente uma refratariedade ao tratamento medicamentoso. Cerca de 75% dos pacientes avaliados em centros terciários, especializados em epilepsias refratárias, apresentam anormalidades à RM do encéfalo(19). Metade dos pacientes epilépticos, na população geral, apresenta anormalidades estruturais detectadas por exame de imagem(20). Contudo, numa série de 341 casos com epilepsia focal e TC de crânio normal, somente 26% dos pacientes não apresentaram alterações à RM do encéfalo(21). Portanto, os exames de imagem, de preferência RM do encéfalo, desempenham papel fundamental na avaliação de pacientes com epilepsia.
5. CRITÉRIOS DE INCLUSÃO
5.1 CLÍNICOS
Serão incluídos neste protocolo de tratamento pacientes com diagnóstico estabelecido de epilepsia, segundo a Classificação Internacional das Epilepsias e Síndromes Epilépticas(10), ou seja, os que tenham apresentado duas crises epilépticas com mais de 24 horas de intervalo.
5.2 EXAMES COMPLEMENTARES EXIGIDOS
Exames de eletroencefalografia em vigília e em sono são obrigatórios para confirmação diagnóstica de epilepsia (diagnóstico diferencial com crises não epilépticas), para detecção de sinais de intoxicação medicamentosa e como auxílio à definição da síndrome epiléptica12. Entretanto, resultado normal do exame não exclui o paciente do protocolo. Em geral (90% dos pacientes com epilepsia), a repetição do exame é suficiente para detectar a alteração. A EEG em sono é obrigatória, tanto pela ativação que este estado fisiológico exerce sobre as descargas epileptiformes (que podem ser ocultadas em exames de vigília), quanto para uma confirmação de ausência de anormalidades nos casos de suspeita de crises não epilépticas ou de potencial suspensão do tratamento (22).
Em casos de pacientes refratários a tratamentos medicamentosos (persistência de crises epilépticas apesar do uso de dois fármacos anticonvulsivantes de primeira linha, em doses adequadas) (9), os seguintes procedimentos são auxiliares na investigação e condução dos casos:
- RM do encéfalo obrigatória para pacientes com epilepsias focais refratárias, para os quais a presença de uma lesão cerebral é forte preditor de refratariedade a tratamento medicamentoso em monoterapia (18);
- diário de registro de crises, importante para a determinação de refratariedade;
- relatório médico, com descrição dos medicamentos e doses máximas previamente empregadas no tratamento; e
- teste psicométrico para casos de efeitos cognitivos negativos provocados pelo uso de medicamentos convencionais.
6. CRITÉRIOS DE EXCLUSÃO
Serão excluídos deste protocolo de tratamento pacientes com diagnóstico duvidoso de epilepsia ou suspeita de crises não epilépticas: pacientes com eventos paroxísticos não epilépticos.
7. CASOS ESPECIAIS
Recomenda-se individualizar o tratamento de acordo com as necessidades específicas dos grupos, conforme o que segue.
7.1 IDOSOS (IDADE ACIMA DE 60 ANOS)
Recomendam-se antiepilépticos não indutores do metabolismo hepático (como a gabapentina e lamotrigina) ao invés de fármacos antiepilépticos indutores enzimáticos clássicos (como a carbamazepina, fenitoína e fenobarbital)(32,33). O escalonamento de dose deve ser lento, e a dose máxima a ser atingida deve ser menor do que a normalmente recomendada para os medicamentos. Nesta população, deve-se tentar evitar o uso de politerapia medicamentosa anticonvulsivante(9).
7.2 CRIANÇAS E ADOLESCENTES (ATÉ 18 ANOS)
Crianças e adolescentes frequentemente sofrem o estresse não apenas das crises mas também das limitações impostas pela doença às suas atividades de lazer e pelos efeitos adversos de fármacos antiepilépticos. A epilepsia mioclônica juvenil (EMJ) inicia na adolescência e é relativamente fácil de controlar desde que sejam evitados fatores precipitantes de crises (como privação de sono, ingestão álcool e má adesão ao tratamento). A EMJ requer tratamento por toda a vida, pois o índice de recorrência de crises após a retirada de fármacos é superior a 90% (9).
7.3 DOENTES PSIQUIÁTRICOS
Depressão e ansiedade são frequentemente subdiagnosticados em pacientes epilépticos, especialmente nos refratários. Nestes pacientes, é seguro utilizar tanto medicamentos inibidores da recaptação sináptica da serotonina como ansiolíticos. Deve-se evitar o tratamento da epilepsia com fenitoína e fenobarbital, que podem induzir transtornos afetivos, e preferir a lamotrigina, que pode ter efeito estabilizador do humor (9).
8. TRATAMENTO
O objetivo do tratamento da epilepsia é propiciar a melhor qualidade de vida possível para o paciente, pelo alcance de um adequado controle de crises, com um mínimo de efeitos adversos.
A determinação do tipo específico de crise e da síndrome epiléptica do paciente é importante, uma vez que os mecanismos de geração e propagação de crise diferem para cada situação, e os fármacos anticonvulsivantes agem por diferentes mecanismos que podem ou não ser favoráveis ao tratamento (23). Os fármacos anticonvulsivantes atuam através de um ou de vários dos seguintes mecanismos: bloqueio de canais de sódio, aumento da inibição gabaérgica, bloqueio de canais de cálcio ou ligação à proteína SV2A da vesícula sináptica (24).
A decisão de iniciar um tratamento anticonvulsivante baseiase fundamentalmente em três critérios: risco de recorrência de crises, consequências da continuação de crises para o paciente e eficácia e efeitos adversos do fármaco escolhido para o tratamento. O risco de recorrência de crises varia de acordo com o tipo de crise e com a síndrome epiléptica do paciente (25), e é maior naqueles com descargas epileptiformes ao EEG, defeitos neurológicos congênitos, crises sintomáticas agudas prévias e lesões cerebrais e em pacientes com paralisia de Todd (26). Incidência de novas crises epilépticas são inaceitáveis para pacientes que necessitam dirigir, continuar empregados ou ser responsáveis por familiares vulneráveis (27). A decisão de iniciar tratamento fica bem mais fortalecida após a ocorrência de 2 ou mais crises epilépticas não provocadas com mais de 24 horas de intervalo.
Até o momento, foram publicados quatro guias oficiais de recomendações (guidelines), baseados em evidências, para o tratamento da epilepsia. Várias discrepâncias significativas entre eles foram constatadas. Por exemplo, a Academia Americana de Neurologia (AAN) recomenda tanto fármacos estabelecidos (carbamazepina, fenitoína, ácido valproico) como novos anticonvulsivantes (lamotrigina, topiramato) para o tratamento de crises focais com ou sem generalizações secundárias(28), enquanto o guia NICE (National Institute for Clinical Excellence), do Reino Unido, propõe que novos fármacos sejam usados neste tipo de crise somente quando o paciente não responder adequadamente aos já estabelecidos(29). O guia SIGN (Scottish Intercollegiate Guidelines Network) apresenta recomendações intermediárias, selecionando dois fármacos da antiga geração e dois novos como monoterapia de primeira linha(25). Entretanto, a revisão sistemática da ILAE(30) concluiu que a melhor evidência disponível não foi suficiente para ser utilizada em recomendações para diagnóstico, monitorização e tratamento de pacientes com epilepsia.
Com relação a ensaios clínicos randomizados (ECRs), existem, até o momento, seis estudos bem delineados(31-36), todos realizados com epilepsias focais. Em geral, a lamotrigina e gabapentina foram mais efetivas do que a carbamazepina em idosos(34,36). Em adultos jovens, a carbamazepina foi mais efetiva do que o fenobarbital, primidona e vigabatrina(31,33), enquanto o ácido valproico teve eficácia comparável à de carbamazepina. Recente ensaio aberto randomizado comparou carbamazepina, gabapentina, lamotrigina, oxcarbazepina e topiramato em epilepsias focais, bem como ácido valproico, lamotrigina e topiramato em epilepsias generalizadas e inclassificáveis (37,38). O estudo concluiu que a lamotrigina é mais efetiva do que a carbamazepina, gabapentina e topiramato como monoterapia de primeira linha para epilepsia focal (37), e o ácido valproico é mais efetivo (eficácia + tolerabilidade) do que o topiramato e mais eficaz do que a lamotrigina nas epilepsias generalizadas e inclassificáveis (38). Entretanto, a revisão sistemática Cochrane conclui pela igualdade de eficácia (50).
As recomendações da ILAE (30), baseadas apenas em evidências de eficácia e efetividade, para escolha de fármacos anticonvulsivantes são as seguintes:
a - adultos com epilepsia focal - carbamazepina, fenitoína e ácido valproico;
b - crianças com epilepsia focal - carbamazepina;
c - idosos com epilepsia focal - lamotrigina e gabapentina;
d - adultos e crianças com crises TCG, crianças com crises de ausência, epilepsia rolândica e epilepsia mioclônica juvenil - nenhuma evidência alcançou níveis A ou B.
Numa revisão sistemática incluindo apenas dois ECRs comparando a oxcarbazepina com a fenitoína, foram estudados 480 pacientes com crises parciais. Os resultados foram controversos: quando utilizados os desfechos "tempo para suspensão do tratamento e tempo para incidência de uma primeira crise", houve vantagem para a oxcarbazepina. Porém, com o desfecho "remissão de crises, de 6 a 12meses", não houve diferença entre os medicamentos39. É evidente a carência de estudos comparando as oxcarbazepina e carbamazepina, este último fármaco normalmente considerado de primeira linha para crises parciais. A igualdade de eficácia foi demonstrada no tratamento de epilepsias focais refratárias em revisão sistemática conduzida por Castillo e colaboradores(40), que avaliou dois ECRs, incluindo 961 pacientes, e encontrou uma razão de chances (RC) para redução de 50% ou mais na frequência de crises de 2,96 (IC95% 2,204,00)(40).
Para as crises generalizadas, o ácido valproico permanece como fármaco de primeira escolha(9). A seleção do fármaco deverá levar em consideração outros fatores além da eficácia, tais como efeitos adversos, especialmente para alguns grupos de pacientes (crianças, mulheres em idade reprodutiva, gestantes e idosos), tolerabilidade individual e facilidade de administração. Especificamente para crises de ausência, uma análise sistemática incluindo cinco pequenos estudos, dos quais apenas um randomizado, comparando a eficácia de etossuximida, ácido valproico, lamotrigina e placebo, não foi suficiente para levantar evidências úteis na prática clínica(41). Mais recentemente, um ECR com 453 crianças com diagnóstico recente de epilepsia do tipo ausência, comparou a eficácia do ácido valproico com a da etossuximida e da lamotrigina. Os resultados mostraram eficácia semelhante para o ácido valproico e a etossuximida e inferior para a lamotrigina(42).
Mesmo utilizando fármacos adequados ao tipo de crise, um controle insatisfatório ocorre em cerca de 15% dos pacientes com epilepsia focal, sendo estes candidatos a tratamento cirúrgico da epilepsia(43).
Em caso de falha do primeiro fármaco, deve-se tentar sempre fazer a substituição gradual por outro, de primeira escolha, mantendo a monoterapia. Em caso de falha na segunda tentativa de tratamento em monoterapia, pode-se tentar a combinação de dois fármacos anticonvulsivantes(44,45). Entretanto, somente há evidências de sinergismo entre o ácido valproico e a lamotrigina quando utilizados em combinação no tratamento de crises focais e generalizadas. Poucos pacientes parecem obter benefício adicional com a associação de mais de dois fármacos. Em um estudo prospectivo, 47% de 470 pacientes em tratamento inicial se beneficiaram com o primeiro fármaco, 13% com o segundo e apenas 3% com associação de dois fármacos. Entretanto, outros autores relatam controle adicional de crises em 10%15% dos pacientes refratários a monoterapia com acréscimo do segundo fármaco(46).
8.1 FÁRMACOS E ESQUEMAS DE ADMINISTRAÇÃO
CARBAMAZEPINA
A Carbamazepina é um iminodibenzil que inibe os disparos neuronais corticais repetitivos, sustentados e de alta frequência através do bloqueio dos canais de sódio voltagem-dependente. Também possui uma discreta ação anticolinérgica.
Sua eficácia foi avaliada em duas revisões sistemáticas(47,48). Tudur e colaboradores(44) compararam carbamazepina e fenobarbital em monoterapia. Em quatro diferentes ensaios, incluindo 684 participantes, o estudo não encontrou diferenças entre esses dois fármacos na remissão de crises por 12 meses, nem no tempo de aparecimento da primeira crise. O fenobarbital é menos tolerado do que a carbamazepina. Gamble e colaboradores48 compararam a carbamazepina com a lamotrigina e encontraram maior eficácia da carbamazepina e melhor tolerância à lamotrigina em epilepsias focais e generalizadas. Esta revisão sistemática estudou cinco ensaios, com 1.384 pacientes.
Indicações
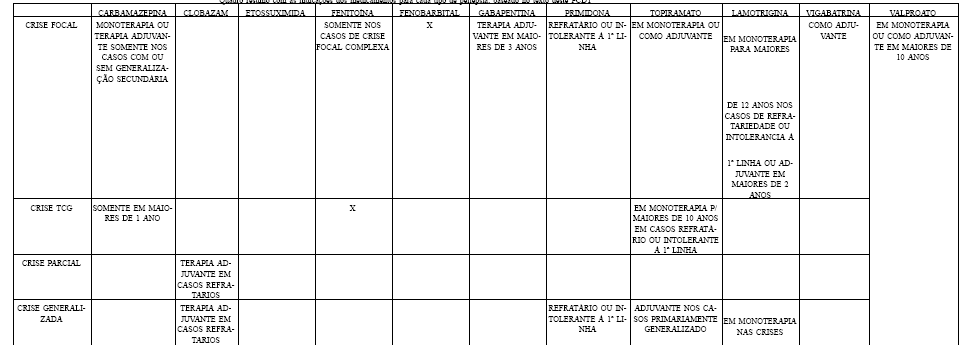
- Monoterapia ou terapia adjuvante de crises focais, com ou sem generalização secundária.
- Crises TCG em pacientes com mais de 1 ano de idade.
CLOBAZAM
O principal sítio de ação dos benzodiazepínicos é um receptor pós-sináptico do ácido gama-aminobutírico (GABA), o principal neurotransmissor inibitório do SNC. Ao ligar-se aos receptores GABAA, o clobazam, como todos os benzodiazepínicos, aumenta a frequência de aberturas destes receptores, aumentando, assim, o índice de correntes inibitórias no cérebro.
O clobazam é rapidamente absorvido pelo trato digestivo, atingindo picos de concentração máximos no sangue em cerca de 90 minutos. A meia-vida é longa (em torno de 20 horas). Este fármaco é fortemente ligado às proteínas séricas (cerca de 85% das moléculas). Suas principais vantagens são a alta eficácia, o rápido início de ação e a boa tolerabilidade. Possíveis desvantagens são o desenvolvimento de tolerância em 40% dos casos e potenciais problemas relacionados à sua retirada (abstinência).
Revisão sistemática conduzida por Michael e Marson(49), incluindo 196 pacientes, concluiu que o clobazam como agente anticonvulsivante adjuvante pode reduzir a frequência de crises nas epilepsias focais. No entanto, o estudo não define que tipo de paciente poderá se beneficiar mais com o fármaco, nem o período de tempo em que o benefício se manterá.
Indicações
- Terapia adjuvante para crises parciais e generalizadas refratárias.
- Terapia intermitente (por exemplo, crises catameniais).
ETOSSUXIMIDA
Este fármaco apresenta um espectro de ação anticonvulsivante bastante restrito, exclusivo para crise de ausência. Seu principal mecanismo de ação é o bloqueio dos canais de cálcio, com consequente inibição do circuito tálamo-cortical, que está intimamente relacionado à geração das crises de ausência.
A etossuximida é útil no tratamento em monoterapia das crises de ausência típicas e como adjuvante nas mioclonias negativas, crises atônicas e mioclonias(41).
Indicações
- Tratamento de crises de ausência em pacientes com ou mais de 3 anos de idade.
- Tratamento adjuvante de mioclonias negativas, crises astáticas e certos tipos de epilepsias mioclônicas.
FENITOÍNA
Seu principal mecanismo de ação é o bloqueio dos canais de sódio voltagem-dependentes, o que lhe confere grande eficácia contra crises epilépticas de início focal.
Após ingestão, a fenitoína atinge picos de concentração em cerca de 6 horas, sendo fortemente ligada às proteínas plasmáticas (mais de 85%), com uma meia-vida de eliminação em torno de 20 horas. Está contraindicada nas crises de ausência e mioclônicas, podendo ser efetiva nas crises tônicas (próprias da síndrome de Lennox-Gastaut)(50-52).
Suas principais desvantagens são efeitos adversos de curto e longo prazos, limitações para uso crônico em mulheres (efeitos estéticos e propriedades teratogênicas) e janela terapêutica restrita e muito próxima dos níveis tóxicos, necessitando de frequentes monitorações dos níveis séricos. Devido à sua farmacocinética peculiar, após atingir dosagens em torno de 300 mg/dia, pequenos incrementos de dose podem gerar aumentos desproporcionais dos níveis séricos, o que exige cautela em sua administração.
Revisões sistemáticas não encontraram diferenças significativas de eficácia entre fenitoína e fenobarbital em monoterapia para crises focais e TCG(50) (apesar de a fenitoína ter sido mais bem tolerada), entre fenitoína e ácido valproico em monoterapia para crises focais e TCG(51) e entre fenitoína e carbamazepina em monoterapia para crises epilépticas(52)).
Indicações
- Tratamento de crises TCG, focais complexas, ou combinação de ambas, em crianças, adolescentes e adultos.
- Prevenção e tratamento de crises epilépticas durante ou após procedimento neurocirúrgico.
FENOBARBITAL
Este fármaco possui largo espectro de ação com efetividade similar à de outros fármacos anticonvulsivantes. É seguro e disponível em apresentações orais e parenterais. Seu principal mecanismo de ação é o prolongamento da abertura dos canais de cloro, dos receptores GABAA e consequente hiperpolarização da membrana pós-sináptica. O fenobarbital também pode bloquear os canais de sódio e potássio, reduzir o influxo de cálcio pré-sináptico e, provavelmente, reduzir as correntes mediadas pelo glutamato.
Apresenta rápida absorção por via oral, porém uma meiavida de eliminação longa (2-7 dias), apesar de ser fracamente ligado às proteínas (20%-50%). As principais desvantagens são seus efeitos colaterais, principalmente na área cognitiva, o que limita seu uso tanto em crianças quanto em idosos. Não é adequado tentar a substituição de fenobarbital em pacientes bem controlados, a menos que seu uso esteja associado a efeitos adversos inaceitáveis. A retirada deve ser feita em dosagens muito pequenas e por longo período de tempo devido ao risco de crises por abstinência. Doses elevadas devem ser evitadas (em adultos, dose máxima de 300 mg/dia).
O fenobarbital ainda é largamente utilizado na prática clínica, por apresentar eficácia equivalente à de fenitoína no tratamento em monoterapia tanto de crises focais como nas generalizadas(53).
Indicação
- Tratamento de crises focais e generalizadas de pacientes de qualquer idade, inclusive recém-nascidos.
GABAPENTINA
A gabapentina apresenta estrutura semelhante à do GABA, no entanto não tem nenhuma interação com os receptores GABAA ou GABAB. Seu sítio de ligação é a proteína alfa2-gama, uma subunidade dos canais de cálcio voltagem-dependentes, embora ainda não haja uma compreensão completa do exato mecanismo anticonvulsivante deste fármaco(54).
Ensaios clínicos testaram sua eficácia apenas com a dose de 2.400 mg/dia, mas, na prática, doses mais elevadas podem ser benéficas(55,56). Em ECR duplo-cego, a gabapentina demonstrou eficácia e tolerabilidade semelhantes às de carbamazepina em monoterapia para o tratamento de epilepsia parcial com ou sem generalização secundária(57,58). No entanto, num estudo aberto comparativo de efetividade (eficácia mais tolerabilidade), a gabapentina mostrou ser inferior a lamotrigina no desfecho "tempo de falha no tratamento" e inferior a carbamazepina no desfecho "tempo de remissão de crises em 12 meses"(37). Em crianças, ela foi avaliada para tratamento adjuvante de crises focais refratárias. Em ECR duplo-cego contra placebo, em crianças de 3-12 anos, a eficácia de gabapentina foi significativa em doses de 23-35 mg/kg/dia(59). Em todos os estudos, houve boa tolerabilidade com baixa toxicidade.
A gabapentina apresenta uma absorção saturável, dependente de dose, ou seja, em doses maiores pode haver menor absorção no duodeno, levando a uma menor eficácia (60). A absorção varia de pessoa para pessoa. Por não ser ligada a proteínas plasmáticas, é eliminada pelos rins, não interferindo com o metabolismo de outros fármacos (61,62), o que a torna ideal para idosos e para pacientes com doença crônica que geralmente usam outros medicamentos (34,48).
Estudos clínicos demonstraram ser ela bem tolerada, não tendo apresentado efeitos adversos significativos. Estudos em crianças indicaram a ocorrência de alguns distúrbios comportamentais, como agressividade e irritabilidade, que parecem ser mais frequentes em crianças com deficiência mental ou com problemas comportamentais prévios (63,64). Sedação, ataxia e ganho de peso também foram relatados. A gabapentina tem poucos efeitos cognitivos, não tendo sido observados efeitos teratogênicos durante a gestação (65,66).
Revisão sistemática realizada por Marson e colaboradores (67), composta por cinco ECRs e incluindo 997 pacientes, concluiu que a gabapentina tem eficácia como agente adjuvante em pacientes com epilepsia focal refratária. No entanto, foi feita a ressalva de que os trabalhos revisados foram de relativa curta duração, deixando, portanto, de mostrar evidências para uma eficácia de longo prazo. Os resultados também não podem ser extrapolados para monoterapia ou para pacientes com outros tipos de epilepsia.
Indicação
- Terapia adjunta para crises focais com ou sem generalização secundária em pacientes com mais de 3 anos de idade.
PRIMIDONA
A primidona, com base em sua estrutura química, não pode ser considerada um barbitúrico; no entanto, parte do seu efeito clínico pode ser atribuído à biotransformação hepática de suas moléculas em fenobarbital.
Um grande estudo multicêntrico controlado comparou 622 pacientes com epilepsia focal, não encontrando qualquer vantagem na eficácia de primidona sobre o fenobarbital, fenitoína e carbamazepina (68). Além de controlar crises focais em um menor número de pacientes, houve grande exclusão de pacientes que faziam uso de primidona devido a seus efeitos sedativos.
Indicação:
- Tratamento de crises focais e generalizadas em pacientes refratários ou intolerantes aos fármacos de primeira linha (69).
TOPIRAMATO
Este fármaco é bem absorvido e minimamente ligado àsproteínas plasmáticas. É parcialmente metabolizado no fígado, e cerca de 60% da dose é excretada inalterada na urina. Seu metabolismo sofre a influência de fármacos indutores de enzimas hepáticas, tendo a meia-vida diminuída com o uso concomitante destes fármacos.
O topiramato apresenta um largo espectro de eficácia, e sua estrutura é distinta da dos outros anticonvulsivantes, tendo sido implicado em vários mecanismos de ação, incluindo bloqueio dos canais de sódio dependentes de voltagem, modulação negativa dos canais de cálcio tipo-L, ativação da condutância do potássio, potencialização da ação inibitória GABAérgica, além de antagonismo a receptores glutamatérgicos e inibição da anidrase carbônica(54).
Revisão sistemática conduzida por Jette e colaboradores(70) confirmou a eficácia do topiramato como fármaco adjuvante no tratamento das epilepsias focais refratárias. Nesse estudo, foram revisados dez ECRs, incluindo 1.312 pacientes. Os estudos foram relativamente de curta duração (11-19 semanas na fase duplo-cega). Comparado ao placebo, o risco relativo (RR) para 50% ou mais de redução de crises foi de 2,85 (IC95% 2,27-3,59). Uma análise de regressão de doses revelou aumento do efeito terapêutico proporcional à dose utilizada, mas nenhuma vantagem adicional com doses acima de 300 mg ou de 400 mg/dia. Ataxia, tonturas, fadiga, náusea, sonolência e "anormalidades do pensamento" são efeitos adversos associados ao topiramato.
Em recente revisão sistemática, Ben-Menachem e colaboradores71 encontraram três estudos randomizados, controlados e duplo-cegos que demonstraram adequada efetividade do topiramato em monoterapia em pacientes com epilepsia recentemente diagnosticada. Os estudos mostraram que o uso de altas doses de topiramato (400500 mg/dia), comparado ao de baixas doses (50 mg/dia), está associado a significativa redução do número de crises após 6 meses de tratamento (54% versus 39%; p = 0,02) e a maior tempo para a ocorrência de uma primeira crise (p < 0,001), além de maior probabilidade de remissão de crises após 12 meses de tratamento (76% versus 59%; p = 0,001). Estes desfechos estiveram diretamente ligados às concentrações plasmáticas de topiramato (71,72). Em estudo comparativo com carbamazepina (600 mg/dia) e com ácido valproico (1.250 mg/dia), não houve diferença significativa na redução de crises em 6 meses de tratamento em relação a topiramato (100 e 200 mg/dia). Os índices de redução de crises se mantiveram entre 44% e 49% com os 3 medicamentos73. Os efeitos adversos mais encontrados com o uso de topiramato durante a fase de escalonamento de doses nos três estudos foram parestesias (25%), fadiga (16%), tonturas (13%), sonolência (13%) e náuseas (10%). Na fase de manutenção, foram observadas cefaleia (20%), diminuição do apetite (11%) e perda de peso (11%)(72-74). Arroyo e colaboradores (73) encontraram disfunção cognitiva em 15% dos pacientes em uso de 50 mg/dia e em 24% dos pacientes em uso de 400 mg/dia. A incidência destes efeitos adversos parece ser menor em crianças e adolescentes (75).
Em estudo comparativo entre topiramato (50-175 mg/dia) e ácido valproico (500- 1.750 mg/dia), em pacientes com epilepsia mioclônica juvenil, Araújo Filho e colaboradores(76) não encontraram diferenças significativas em 11 de 13 subtestes neuropsicológicos (WISC III) entre os grupos. No entanto, o ácido valproico foi associado a escores significativamente maiores em testes de memória de curto prazo, atenção e velocidade de processamento, quando comparado com topiramato. Outro efeito adverso frequente observado com o uso de topiramato foi nefrolitíase (15%).
Recente estudo aberto randomizado demonstrou que a eficácia do topiramato em adultos e crianças é equivalente à de carbamazepina nas epilepsias focais e à de ácido valproico nas epilepsias generalizadas recentemente diagnosticadas. No entanto, o estudo teve uma série de limitações referentes ao não cegamento, não controle de doses utilizadas e a não classificação adequada dos tipos de crises (33,38). O topiramato é altamente eficaz no tratamento de pacientes com síndrome de Lennox-Gastaut bem como no de pacientes com epilepsias catastróficas da infância (77).
Indicações:
- Monoterapia para crises focais ou primariamente TCGs em pacientes mais de 10 anos de idade com intolerância ou refratariedade a outros medicamentos de primeira linha.
- Terapia adjuvante para crises focais, primariamente generalizadas ou crises associadas com a síndrome de Lennox-Gastaut em pacientes mais de 2 anos de idade.
LAMOTRIGINA
O principal mecanismo de ação da lamotrigina parece envolver a inibição dos canais de sódio voltagem-dependentes, resultando em inibição dos potenciais elétricos pós-sinápticos. Não parece ter efeito GABAérgico e não tem semelhança química com os anticonvulsivantes indutores enzimáticos (fenobarbital, fenitoína, carbamazepina) (78).
Recente ECR aberto indicou a lamotrigina como fármaco de primeira escolha para epilepsia focal, por ter tido eficácia equivalente mas ter sido mais bem toleradado do que o ácido valproico (32). Porém, no mesmo estudo, a lamotrigina foi menos eficaz do que o ácido valproico nas epilepsias generalizadas e inclassificáveis (33). Alguns autores sugerem uma associação de lamotrigina com ácido valproico em pacientes refratários, a fim de obter uma eficácia maior, devido às possíveis interações farmacodinâmicas favoráveis entre os dois medicamentos (31). Outros estudos, ainda, demonstraram que a lamotrigina é mais bem tolerada do que a carbamazepina em idosos (34,79).
Uma atualização dos parâmetros práticos recomendados pela ILAE no tratamento da epilepsia em mulheres, com foco na gestação, realizou uma revisão sistemática de artigos publicados entre 1985 e 2007. Conclui-se que é altamente provável que a exposição intrauterina a ácido valproico, no primeiro trimestre da gestação, tenha maior risco para malformações congênitas importantes em relação a carbamazepina, e possivelmente em comparação com a fenitoína e com a lamotrigina. No entanto, convém lembrar que o uso de anticoncepcionais orais diminui a concentração plasmática de lamotrigina, além do que, durante a gestação, o metabolismo deste fármaco encontra-se aumentado (80). Recente estudo demonstrou que, com uma rigorosa monitorização do paciente, o risco de aumento da frequência de crises não foi maior do que com outros anticonvulsivantes (81). Por conta dos menores riscos de teratogênese e por proporcionar menor ganho de peso em relação ao ácido valproico, lamotrigina tem sido apontada como um medicamento de escolha no tratamento da epilepsia mioclônica juvenil em mulheres em idade fértil (82). Porém nem todos os tipos de crises são tratados com a mesma eficácia por lamotrigina, e alguns deles inclusive podem ser agravados, como determinadas crises mioclônicas (83).
Revisão sistemática da Cochrane, recentemente revisada, reafirmou a eficácia da lamotrigina como terapia adjuvante na redução da frequência de crises em pacientes com epilepsias focais refratárias84. Foram revisados 1.243 pacientes em três estudos de lamotrigina como fármaco adjuvante e em oito estudos cruzados. Comparada ao placebo, a lamotrigina apresentou maior redução na frequência de crises (50% ou mais), com uma RC geral de 2,71 (IC95% 1,87-3,91).
Indicações:
- Monoterapia para crises focais com ou sem generalização secundária em pacientes com mais de 12 anos de idade em situações de intolerância ou refratariedade a medicamentos de primeira linha.
- Monoterapia para crises primariamente generalizadas em pacientes com mais de 12 anos de idade em situações de intolerância ou refratariedade a medicamentos de primeira linha.
- Terapia adjuvante para crises focais em pacientes mais de 2 anos de idade.
- Terapia adjuvante para crises generalizadas da síndrome de Lennox-Gastaut em pacientes com mais de 2 anos de idade.
VIGABATRINA
A vigabatrina é um análogo estrutural do ácido gama-aminobutírico (GABA) que inibe irreversivelmente a GABA-transaminase (GABA-T), aumentando os níveis sinápticos de GABA no cérebro(85).
As duas maiores indicações clínicas deste fármaco são o tratamento de crises do tipo espasmos infantis e de crises focais refratárias. Seu uso em adultos restringe-se a pacientes com epilepsia grave que não respondem a outros medicamentos anticonvulsivantes devido a seus potenciais graves efeitos adversos (86). No entanto, comparada a carbamazepina, sua eficácia é inferior em pacientes com epilepsia recentemente diagnosticada (87). Ela também agrava mioclonias (88). Em crianças, no entanto, a vigabatrina é altamente efetiva na síndrome de West, especialmente quando associada a esclerose tuberosa (89).
Numa revisão sistemática, incluindo 747 pacientes em uso adjuvante de vigabatrina para tratamento de epilepsias focais, em 11 ECRs, Hamming e colaboradores (90) concluíram que a vigabatrina é 2,5 vezes mais eficaz do que placebo. Também os pacientes tratados com vigabatrina apresentaram 2,5 vezes mais efeitos adversos quando comparados a placebo (principalmente fadiga e sonolência). A eficácia da vigabatrina é maior nas crises focais sem generalização secundária, tanto como medicamento adicional (91,92) quanto em monoterapia (93,94).
A vigabatrina é eficaz para tratamento da síndrome de West (espasmos infantis, hipsarritmia e retardo do desenvolvimento neuropsicomotor). Apesar de haver poucos estudos metodologicamente aceitáveis e, até o momento, poucos pacientes selecionados, Hancock e colaboradores (89) concluíram, numa revisão sistemática, que a vigabatrina deve ser considerada o medicamento de primeira escolha em espasmos infantis associados a esclerose tuberosa.
A vigabatrina piora crises generalizadas primárias, provocaaumento de ausências e pode desencadear crises mioclônicas. É contraindicada para epilepsias mioclônicas e para crises TCG primárias (83). Os principais efeitos adversos são irritabilidade, insônia e distúrbios psiquiátricos (85). Efeitos sobre os campos visuais (retração concêntrica) foram confirmados em muitos estudos, sendo encontrados em até 40% dos pacientes tratados (28). Eles são progressivos nos pacientes que continuam usando o medicamento e dependem de dose cumulativa, sendo reversível apenas quando suspensa precocemente. Estes efeitos são provavelmente resultantes da toxicidade da vigabatrina sobre os cones da retina periférica e devem ser ativamente buscados através de estudos de eletrorretinografia e potenciais evocados visuais, uma vez que a maioria dos pacientes é assintomática (28).
Indicações:
- Monoterapia no tratamento de espasmos infantis.
- Terapia adjunta para crises focais com ou sem generalização secundária em pacientes de qualquer idade.
PRECURSORES DO ÍON VALPROATO: ÁCIDO VALPROICO E VALPROATO DE SÓDIO
Valproato é o íon circulante no sangue responsável pelo efeito anticonvulsivante das diferentes formulações farmacêuticas. Foi inicialmente comercializado sob a forma ácida e depois na de sal (de sódio ou de magnésio) e de amido. Mais recentemente, foi desenvolvida a molécula de divalproato de sódio. Não há na literatura ECR que tenha demonstrado superioridade em eficácia anticonvulsivante entre as diferentes formulações.
O ácido valproico é um dos principais anticonvulsivantes utilizados, com eficácia estabelecida para múltiplos tipos de crises. Picos máximos de concentração são atingidos 2 horas após a ingestãooral. É altamente ligado às proteínas (90%), e a meia-vida de eliminação é de cerca de 15 horas.
Seu mecanismo de ação pode envolver redução na frequência de disparos dos canais de sódio, ativação da condutância do potássioe, possivelmente, ação direta sobre outros canais iônicos. É sabido que o ácido valproico tem um efeito GABAérgico através da elevação do GABA cerebral por diversos mecanismos: inibição da GABA-transaminase, aumento das enzimas sintetizadoras do GABA, aumento da liberação e inibição da recaptação do GABA.
Suas principais desvantagens são maior incidência de efeitos adversos em mulheres (alterações hormonais, ganho de peso e teratogenicidade) e em crianças com menos de 2 anos de idade, especialmente naquelas em politerapia, com doenças metabólicas congênitas ou com retardo mental (devido a risco aumentado de desenvolvimento de hepatotoxicidade fatal). O uso de ácido valproico para crises focais apresenta eficácia limitada devido principalmente à necessidade de doses significativamente maiores do que as usadas para crises generalizadas.
Revisão sistemática, incluindo cinco ensaios randomizados e 1.265 pacientes, não encontrou evidências para apoiar o uso de carbamazepina em crises focais nem de ácido valproico em crises generalizadas95. No entanto, os intervalos de confiança obtidos foram muito amplos para confirmar equivalência entre os dois fármacos nos diferentes tipos de crises epilépticas. Assim, na ausência de evidência definitiva, continuam a ser adotados critérios tradicionais de tratamento.
Indicação:
- Monoterapia e terapia adjunta de pacientes com mais de 10 anos de idade e com qualquer forma de epilepsia.
8.2 FÁRMACOS E ESQUEMAS DE ADMINISTRAÇÃO
Carbamazepina:
comprimidos de 200 mg, xarope de 20 mg/ml
Dose inicial:
- Adultos: 200 mg/dia
- Crianças de 6-12 anos: 100 mg/dia
- Crianças abaixo de 6 anos: 5-10 mg/kg/dia
Escalonamento:
- Adultos: 200 mg/dia/semana
- Crianças de 6-12 anos: 100 mg/dia/semana
- Crianças < 6 anos: 5-10 mg/kg/dia/semana
Dose máxima:
- Adultos: 1.800 mg/dia
- Crianças de 6 a 12 anos: 600-1.000 mg/dia
- Crianças < 6 anos: 35 mg/kg/dia
Intervalo de dose: 2 a 3 administrações/dia
Clobazam: comprimidos de 10 e 20 mg
Dose inicial: 5-10 mg/dia
Escalonamento: 5 mg/dia/semana
Dose máxima: 40 mg/dia
Intervalo de dose: 1 administração/dia (à noite)
Etossuximida: xarope de 50 mg/ml
Dose inicial: 250 mg/dia
Escalonamento: 250 mg/dia/semana
Dose máxima: 1.500 mg/dia
Intervalo de dose: 2-3 administrações/dia
Fenitoína: comprimidos de 100 mg, suspensão oral 25 mg/ml
Dose inicial: 100 mg/dia
Escalonamento: 100 mg/dia/semana
Dose máxima: 500 mg/dia
Intervalo de dose: 1-2 administrações/dia
Fenobarbital: comprimidos de 100 mg e solução oral 40 mg/ml
Dose inicial: 50 mg/dia
Escalonamento: 50 mg/dia/semana
Dose máxima: 300 mg/dia
Intervalo de dose: dose única diária
Primidona: comprimidos de 100 e 250 mg
Dose inicial: 100 mg/dia
Escalonamento: 100 mg/dia/semana
Dose máxima: 750 mg/dia
Intervalo de dose: 3 administrações/dia
Ácido valproico: comprimidos ou cápsulas de 250 mg, comprimidos de 500 mg e solução e xarope de 50 mg/ml.
Dose inicial: 250 mg/dia
Escalonamento: 250 mg/dia a cada 3 dias
Dose máxima: 3.000 mg/dia
Intervalo de dose: 2 administrações/dia
Gabapentina: cápsulas de 300 e 400 mg
Dose inicial: 15 mg/kg/dia ou máximo de 300 mg/dia
Escalonamento: 300 mg/dia (15 mg/kg/dia)
Dose máxima: 3.600 mg/dia (50-100 mg/kg/dia)
Intervalo de dose: 3 administrações/dia
Topiramato: comprimidos 25, 50 e 100 mg
- Adultos:
Dose inicial: 25 mg/dia
Escalonamento: 25-50 mg/semana
Dose máxima: 400 mg/dia
Intervalo de dose: 2 administrações/dia
- Crianças de 6-16 anos:
Dose inicial: 1-3 mg/kg/dia
Escalonamento: 1-3 mg/kg/semana
Dose máxima: 9 mg/kg/dia
Intervalo de dose: 2 administrações/dia
- Crianças de 2-6 anos:
Dose inicial: 0,5-1 mg/kg/dia
Escalonamento: 1-3 mg/kg/semana
Dose máxima: 9 mg/kg/dia
Intervalo de dose: 2 administrações/dia
Obs.: Pacientes com insuficiência renal, recomenda-se utilizar a metade da dose.
Lamotrigina: comprimidos 25, 50 e 100 mg
Monoterapia:
Dose inicial: 25 mg/dia por 2 semanas; 50 mg/dia por mais 2 semanas
Escalonamento: 50-100 mg a cada 1-2 semanas
Dose máxima: 500 mg/dia (1-5 mg/kg/dia) Intervalo de dose: 1 a 2 administrações/dia
- Terapia adjuvante com ácido valproico:
Dose inicial: 25 mg a cada 2 dias por 2 semanas (0,15 mg/kg/dia); 25 mg/dia por mais 2 semanas (0,3 mg/kg/dia)
Escalonamento: 25-50 mg a cada 1-2 semanas (0,3 mg/kg)
Dose máxima: 500 mg/dia (1-5 mg/kg/dia)
Intervalo de dose: 1 a 2 administrações/dia
- Terapia adjuvante com fármacos anticonvulsivantes indutores enzimáticos
Dose inicial: 50 mg/dia por 2 semanas (0,6 mg/kg/dia); 100 mg/dia por mais 2 semanas (1,2 mg/kg/dia)
Escalonamento: 100 mg a cada 1-2 semanas (1,2 mg/kg)
Dose máxima: 700 mg/dia (5-15 mg/dia)
Intervalo de dose: 2 administrações/dia
Vigabatrina: comprimidos de 500 mg
Dose inicial: 500 mg/dia
Escalonamento: 500 mg/semana
Dose máxima: 3.000 mg/dia (150-200 mg/kg/dia)
Intervalo de dose: 1 a 2 administrações/dia
8.3 COMBINAÇÕES DE FÁRMACOS (POLITERAPIA)
Há evidências de sinergismo entre o ácido valproico e a lamotrigina quando utilizados em combinação no tratamento de crises focais e generalizadas(96,97). Outras combinações possíveis carecem de evidências(98-101). Há, também, evidências de que o uso de carbamazepina em combinação com lamotrigina pode favorecer o aparecimento de efeitos adversos neurotóxicos devido a interações farmacodinâmicas adversas(102).
8.3.1 CRITÉRIOS PARA O USO DE ASSOCIAÇÃO MEDICAMENTOSA (103)
- Controle inadequado de crises com duas monoterapias seqüenciais; ou
- Paciente de alto risco para agravamento de crises, definido por epilepsias generalizadas sintomáticas, quando em uso de anticonvulsivante de espectro estreito.
8.3.2 CRITÉRIOS PARA TROCA DE MEDICAMENTO (MANUTENÇÃO DE MONOTERAPIA):
Assegurada adesão ou nível sérico (quando disponível) recomenda-se a troca de medicamentos nas seguintes situações:
- Intolerância a primeira monoterapia em doses adequadas; ou
- Falha no controle ou presença de exacerbação de crises.
8.4 TEMPO DE TRATAMENTO - CRITÉRIOS DE INTERRUPÇÃO
POR FALHA DE TRATAMENTO
O período de avaliação da resposta será de 3 meses com o tratamento em doses máximas, após o que, caso não haja resposta, um segundo medicamento será adicionado ao esquema terapêutico.
POR REMISSÃO DAS CRISES
O paciente é considerado livre de crises quando elas não ocorrerem por pelo menos 2 anos, em vigência de tratamento com dose inalterada neste período. Pacientes com crises após este período são considerados refratários (4). Estima-se que 30% sejam refratários aos fármacos atuais.
Não há diretrizes definitivas para a interrupção do tratamento. A decisão deve ser tomada individualmente, considerando-se as consequências médicas e psicossociais da recorrência dos ataques e os riscos de efeitos adversos do tratamento prolongado. Nas epilepsias sintomáticas, a persistência das crises está definida pela persistência da lesão determinante. Nas epilepsias focais complexas associadas a esclerose temporal mesial, apenas 10% dos pacientes ficam livres de crises contra cerca de 60% com bons resultados cirúrgicos104.
O índice de não recorrência de crise após a suspensão do medicamento é maior nas epilepsias idiopáticas. A persistência de alterações paroxísticas ao EEG está associada a maior risco de recorrência. A maioria dos centros considera um período de tempo mínimo de 3 anos após a última crise associado à ausência de alterações paroxísticas ao EEG para a suspensão do tratamento.
Revisão sistemática, que incluiu sete ECRs com 924 crianças (não houve estudo elegível com adultos), comparou os riscos de recorrência de crises epilépticas após a retirada precoce (menos de 2 anos de remissão de crises) e tardia (mais de 2 anos sem crises) dos anticonvulsivantes105. Retirada precoce de anticonvulsivantes associou-se a maiores índices de recorrência de crises em pacientes com crises focais (RR 1,52; IC (95%): 0,95-2,41) ou ao EEG anormal (RR 1,67; IC(95%): 0,93-3,00).
Portanto, há evidências apoiando uma espera de pelo menos 2 anos livres de crises antes da retirada do medicamento anticonvulsivante em crianças, principalmente se o paciente tiver crises focais ou EEG anormal. Não há evidências para determinar quando suspender o tratamento em crianças e adolescentes com crises generalizadas nem em adultos livres de crises.
9. MONITORIZAÇÃO
O tempo de tratamento da epilepsia é, em geral, imprevisível. Há duas situações em que ele pode ser interrompido: por falha do tratamento ou por remissão completa das crises. O período de reavaliação é de 3 meses. Na reavaliação, o médico verificará eficácia e segurança do tratamento.
A resposta ao tratamento deve ser avaliada com base na redução do número de crises - diário de crises -, bem como na tolerabilidade, levando em consideração os efeitos adversos, especialmente os cognitivos e comportamentais. Sugere-se elaboração de um diário de crises contendo doses do medicamento em uso, descrição das crises e efeitos colaterais.
EFEITOS POSITIVOS
O alvo principal do tratamento da epilepsia é assegurar a melhor qualidade de vida, compatível com a natureza do transtorno epiléptico do paciente e com quaisquer deficiências físicas ou mentais associadas. Para atingi-lo, vários objetivos devem ser buscados. O primeiro deles, e certamente o mais importante, é o controle completo das crises. Fármacos antiepilépticos podem produzir efeitos adversos graves, especialmente quando utilizados em doses elevadas ou em combinação com outros fármacos. Sempre que o controle completo de crises se revelar inalcançável, uma estratégia alternativa adequada é combinar uma frequência de crises mínima desejável com efeitos adversos mantidos dentro de limites aceitáveis. Por exemplo, em pacientes com vários tipos de crise, como na síndrome de Lennox-Gastaut, é importante evitar as crises com maior impacto sobre a qualidade de vida do paciente. Assim, é muito mais importante tentar suprimir as crises atônicas (que levam a quedas fulminantes do pacientes) do que as crises focais ou de ausência atípicas que acompanham o quadro. Da mesma forma, o tratamento das crises TCG exerce maior impacto sobre a qualidade de vida do paciente do que o tratamento das crises focais simples.
Mesmo com um tratamento farmacológico adequado, é importante que o paciente identifique e evite situações que aumentem sua suscetibilidade a crises, como exposição a flashes de luz intermitentes (por exemplo, videogame), privação de sono ou abuso de bebidas alcoólicas.
Níveis terapêuticos, medidos na corrente sanguínea, foram estabelecidos para os fármacos anticonvulsivantes. No Brasil, dispõese de níveis séricos para carbamazepina (níveis terapêuticos entre 412 g/ml), fenitoína (10-20 g/ml), fenobarbital (10-30 g/ml) e ácido valproico (50- 100 g/ml). Eles representam as faixas de concentração dentro das quais a maioria dos pacientes apresenta controle de crises sem efeitos adversos (106). Recomendam-se medidas da concentração sérica dos fármacos anticonvulsivantes, podendo ser úteis nas seguintes situações clínicas (90): 1) avaliar adesão ao tratamento; 2) diagnosticar intoxicação medicamentosa; 3) estabelecer concentrações clinicamente terapêuticas individuais para cada paciente; 4) orientar ajuste de doses quando houver variabilidade farmacocinética (troca de formulação, crianças, idosos, presença de comorbidades); 5) apresentar potenciais alterações farmacocinéticas (gestação, politerapia); e 6) apresentar farmacocinética dose-dependente ou janela terapêutica restrita (por exemplo, fenitoína).
EFEITOS ADVERSOS
Efeitos adversos relacionados ao uso de fármacos antiepilépticos podem ser relacionados ou não à dose. Em geral, os efeitos relacionados à dose utilizada, como letargia, sonolência, ataxia e diplopia, são reversíveis, isto é, desaparecem com a redução da dose ou com a suspensão do fármaco causador dos sintomas. No entanto, alguns quadros provocados por superdosagem são potencialmente graves e irreversíveis, como a síndrome de Stevens-Johnson, observada pela combinação de ácido valproico e lamotrigina. Da mesma forma, reações não relacionadas à dose requerem suspensão imediata do fármaco. Para evitar quadros clínicos graves e de difícil condução, potencialmente fatais, devem ser identificados pacientes pertencentes a grupos de risco para o desenvolvimento de efeitos adversos, especialmente aqueles com história familiar de graves reações alérgicas, idosos, pacientes com massa corporal baixa e com doenças coexistentes (em uso de vários medicamentos).
Na pós-menopausa, artralgias e dores musculares podem indicar osteoporose associada ao uso de fármacos antiepilépticos, especialmente de indutores enzimáticos(9). Nos homens, disfunção sexual e aumento de peso durante tratamento com fármacos antiepilépticos podem ocorrer.
Os principais efeitos adversos relatados encontram-se a seguir arrolados.
- Fenitoína: ataxia, sonolência, letargia, sedação e encefalopatia (dose-dependentes), hiperplasia gengival, hirsutismo e dismorfismo facial (uso crônico).
- Fenobarbital: tontura, sedação, depressão, transtornos comportamentais, prejuízo cognitivo e da concentração, hiperatividade em crianças.
- Clobazam: sonolência, efeitos cognitivos e compotamentais, desenvolvimento de tolerância
- Primidona: semelhantes aos do fenobarbital.
- Carbamazepina: sedação, cefaleia, diplopia, visão turva, rash cutâneo, transtornos gastrointestinais, ataxia, tremor, impotência, hiponatremia, neutropenia.
- Topiramato: sonolência, anorexia, fadiga, nervosismo, pensamento lento, dificuldade de encontrar paalavras, dificuldade de concentração, perda de peso, parestesias, dores abdominais, acidose metabólica, nefrolitíase, miopia e glaucoma de ângulo fechado.
- Gabapentina: aumento do apetite, ganho de peso, tontura, ataxia, nistagmo, cefaleia, tremor, fadiga, diplopia, náusea, comportamento agressivo em crianças.
- Lamotrigina: rash cutâneo, cefaleia, diplopia, náusea, tontura, ataxia, tremor, astenia e ansiedade.
- Etossuximida: transtornos gastrointestinais, sonolência, perda de peso, fotofobia, euforia, soluços, cefaleia, transtornos comportamentais (menos frequentes).
- Ácido valproico: sonolência, fadiga, tremor (relacionados ao SNC); insuficiência hepática, pancreatite hemorrágica aguda, encefalopatia hiperamonêmica, trombocitopenia, ganho de peso, alopecia (sistêmicos).
- Vigabatrina: defeitos no campo visual, sedação, cefaleia, tontura, ataxia, transtornos de memória e comportamentais, parestesias, ganho de peso e tremor.
10 Regulação/Controle/Avaliação Pelo Gestor
Os pacientes com epilepsia refratária devem ser atendidos por médicos especialistas em Neurologia em hospitais terciários, habilitados na alta complexidade em Neurologia/Neurocirurgia.
Devem ser observados os critérios de inclusão e exclusão de pacientes neste protocolo, a duração e a monitorização do tratamento, bem como a verificação periódica das doses prescritas e dispensadas e a adequação de uso de medicamento.
11. TERMO DE ESCLARECIMENTO E RESPONSABILIDADE - TER
É obrigatória a informação ao paciente ou a seu responsável legal dos potenciais riscos, benefícios e efeitos adversos relacionados ao uso dos medicamentos preconizados neste protocolo. O TER é obrigatório ao se prescrever medicamento do Componente Especializado da Assistência Farmacêutica.
12. REFERÊNCIAS BIBLIOGRÁFICAS
1. Engel J Jr, Pedley TA. Introduction: what is epilepsy? In: Engel J Jr, Pedley TA, eds. Epilepsy: a comprehensive textbook, 2nd edition. Philadelphia: Lippincott Williams & Wilkins; 2008: 1-13.
2. Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, Engel J Jr. Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005; 46: 470-2
3. Banerjee PN, Hauser WA. Incidence and prevalence. In: Engel Jr J, Pedley TA, eds. Epilepsy: a comprehensive textbook, 2nd edition. Philadelphia: Lippincott Williams & Wilkins; 2008: 45-56.
4. Kwan P, Brodie M. Early identification of refractory epilepsy. N Engl J Med 2000; 342: 314-9.
5. Kwan P, Sander JW. The natural history of epilepsy: an epidemiological view. J Neurol Neurosurg Psychiatry 2004;75:1376-81.
6. Marino R Jr, Cukiert A, Pinho E. Epidemiological aspects of epilepsy in São Paulo: a prevalence study. Arq. Neuro-Psiquiatr. 1986; 44: 243-54.
7. Fernandes JG, Schmidt MI, Monte TL, Tozzi S, Sander JWAS. Prevalence of epilepsy. The Porto Alegre Study. Epilepsia 1992; 33 (Supl 3): 132.
8. Commmission on Classificationand Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 1981; 22: 489-501.
9. Elger CE, Schmidt D. Modern management of epilepsy: a practical approach. Epilepsy Behav 2008; 12: 501-39.
10. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989; 30: 389-99.
11. Cascino GD. Neuroimaging in epilepsy: diagnostic strategies in partial epilepsies. Semin Neurol 2008; 28: 523-32.
12. Nabbout R, Dulac O. Epileptic syndromes in infancy and childhood. Curr Opin Neurol 2008; 21(2): 161-6.
13. Panayiotopoulos CP, Michael M, Sanders S, Valeta T, Koutroumanidis M. Benign childhood focal epilepsies: assessment of established and newly recognized syndromes. Brain. 2008; 131(Pt 9): 2264-86.
14. Dubé CM, Brewster AL, Baram TZ. Febrile seizures: mechanisms and relationship to epilepsy. Brain Dev. 2009; 31(5): 366-71.
15. Riikonen R. The latest on infantile spasms. Curr Opin Neurol. 2005; 18(2): 91-5.
16. Zupanc ML. Clinical evaluation and diagnosis of severe epilepsy syndromes of early childhood. J Child Neurol. 2009; 24(8 Suppl): 6S-14S.
17. Noachtar S, Rémi J. The role of EEG in epilepsy: a critical review. Epilepsy Behav 2009; 15: 22-33.
18. Koepp M, Woermann FG. Imaging structure and function in refractory focal epilepsy. Lancet Neurol 2005; 4: 42-53.
19. Li LM, Fish DR, Sisodiya SM, Shorvon SD, Alsanjari N, Stevens JM. High resolution magnetic resonance imaging in adults with partial or secondary generalised epilepsy attending a tertiary referral unit. J Neurol Neurosurg Psychiatry 1995; 59: 384-7.
20. Liu RS, Lemieux L, Bell GS, Hammers A, Sisodiya SM, Bartlett PA, Shorvon SD, Sander JW, Duncan JS. Progressive neocortical damage in epilepsy. Ann Neurol 2003; 53: 312-24.
21. Shorvon SD. A history of neuroimaging in epilepsy 1909-2009. Epilepsia 2009; 50 (Suppl. 3): 39-49.
22. Mendez OE, Brenner RP. Increasing the yield of EEG. J Clin Neurophysiol; 23: 282-93.
23. Perucca E. An introduction to antiepileptic drugs. Epilepsia 2005; 46 (Suppl 4): 31-7.
24. Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci 2004; 5:1-13.
25. Scottish Intercollegiate Guidelines Network. Diagnosis and management of epilepsy in adults: a national clinical guideline. 2006. www.sign.ac.uk. Acessado em 13-3-2010.
26. Dulac O, Leppik IE, Chadwick DW et al. Starting and stopping treatment. In: Engel J Jr, Pedley TA eds. Epilepsy: a comprehensive textbook. 2nd edition. Philadelphia: Wolter Kluwer/Lippincott Williams and Wilkins; 2008. p. 1301-9.
27. Stephen LJ, Brodie MJ. The management of a first seizure. Still a major debate. Speccial problems: adults and elderly. Epilepsia 2008; 49 (Suppl. 1): 45-9.
28. French J, Kanner AM, Bautista J, Abou-Khalil B, Browne T, Harden CL, Theodore WH et al. Efficacy and tolerability of the new antiepileptic drugs. I: Treatment of new onset epilepsy: report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2004; 62: 1252-60.
29. National Institute for Clinical Excellence. The epilepsies: the diagnosis and management of the epilepsies in adults and children in primary and secondary care. (www.nice.org.uk/CG020NICEguideline). 2004. Acessado em 11-4-2009.
30. Glauser T, Ben-Menachen E, Bourgeois B, Cnaan A, Chadwick D, Guerreiro C et al. ILAE treatment guidelines: evidence-based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 2006; 47: 1094-1120.
31. Mattson RH, Cramer JA, Collins JF, Smith DB, Delgado-Escueta AV, Browne TR, Williamson PD, Treiman DM, McNamara JO, McCutchen CB et al. Comparision of carbamazepine, phenobarbital, phenytoin, and primidone in partial and secondarily generalized tonic-clonic seizures. N Engl J Med 1985; 313: 145-51.
32. Guerreiro MM, Vigonius U, Pohlmann H, de Manreza ML, Fejerman N, Antoniuk SA, Moore A. A double-blind controlled clinical trial of oxcarbamazepine versus phenytoin in children and adolescents with epilepsy. Epilepsy Res 1997; 27: 205-13.
33. Chadwick D. Safety and efficacy of vigabatrin and carbamazepine in newly diagnosed epilepsy: a multicentre randomised double-blind study. Vigabatrin European Monotherapy Study Group. Lancet 1999; 354: 13-9.
34. Rowan AJ, Ramsay RE, Collins JF, Pryor F, Boardman KD, Uthman BM, Spitz M, Frederik T, Towne A, Carter GS, Marks W, Felicetta J, Tomyanovich ML, VA Cooperative Study 428 Group. New onset geriatric epilepsy: a randomized study of gabapentin, lamotrigine, and carbamazepine. Neurology 2005; 64: 1868-73.
35. Mattson RH, Cramer JA, Collins JF. A comparision of valproate with carbamazepine for the treatment of complex partial seizures and secondarily generalized tonic-clonic seizures in adults. The Department of Veterans Affairs Epilepsy Cooperative Study No. 264 Group. N Engl J Med 1992; 327: 765-71.
36. Brodie MJ, Overstall PW, Giorgi L. Multicentre, double-blind, randomised comparision between lamotrigine and carbamazepine in elderly patients with newly diagnosed epilepsy. The UK Lamotrigine Elderly Study Group. Epilepsy Res 1999; 37: 81-7.
37. Marson AG, Al-Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW. The SANAD study of effectiveness of carbamazepine, gabapentine, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet 2007; 369: 1000-15.
38. Marson AG, Al-Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet 2007; 369: 1016-26.
39. Muller M, Marson AG, Williamson PR. Oxcarbazepine versus phenytoin monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2006, Issue 2. Art. No.: CD003615. DOI: 10.1002/14651858.CD003615.pub2.
40. Castillo SM, Schmidt DB, White S, Shukralla A. Oxcarbazepine add-on for drug-resistant partial epilepsy. Cochrane Database of Systematic Reviews 2000, Issue 3. Art. No.: CD002028. DOI: 10.1002/14651858.CD002028.
41. Posner EB, Mohamed KK, Marson AG. Ethosuximide, sodium valproate or lamotrigine for absence seizures in children and adolescents. Cochrane Database of Systematic Reviews 2005, Issue 4. Art. No.: CD003032. DOI: 10.1002/14651858.CD003032.pub2.
42. Glauser TA, Cnaan A, Shinnar S, Hirtz DG, Dlugos D, Masur D, Clark PO, Capparelli EV, Adamson PC; Childhood Absence Epilepsy Study Group. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med 2010; 362(9): 790-9.
43. Duncan JS, Sander JW, Sisodiya SM, Walker MC. Adult epilepsy. Lancet 2006; 367: 1087-100.
44. Kwan P, Brodie MJ. Combination therapy in epilepsy: when and what to use. Drugs 2006; 66: 1817-29.
45. French JA, Faught E. Rational polytherapy. Epilepsia 2009; 50 (Suppl 8): 63-8.
46. Elger CE, Fernández G. Options after the first antiepileptic drug has failed. Epilepsia 1999; 40 (Suppl 6): S9-S12.
47. Tudur Smith C, Marson AG, Williamson PR. Carbamazepine versus phenobarbitone monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2003, Issue 1. Art. No.: CD001904. DOI: 10.1002/14651858.CD001904.
48. Gamble CL, Williamson PR, Marson AG. Lamotrigine versus carbamazepine monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2006, Issue 1. Art. No.: CD001031. DOI: 10.1002/14651858.CD001031.pub2.
49. Michael B, Marson AG. Clobazam as an add-on in the management of refractory epilepsy. Cochrane Database of Systematic Reviews 2008, Issue 2. Art. No.: CD004154. DOI: 10.1002/14651858.CD004154.pub4.
50. Taylor S, Tudur Smith C, Williamson PR, Marson AG. Phenobarbitone versus phenytoin monotherapy for partial onset seizures and generalized onset tonic-clonic seizures. Cochrane Database of Systematic Reviews 2003, Issue 2. Art. No.: CD002217. DOI: 10.1002/14651858.CD002217.
51. Tudur Smith C, Marson AG, Williamson PR. Phenytoin versus valproate monotherapy for partial onset seizures and generalized onset tonic-clonic seizures. Cochrane Database of Systematic Reviews 2001, Issue 4. Art. No.: CD001769. DOI: 10.1002/14651858.CD001769.
52. Tudur Smith C, Marson AG, Clough HE, Williamson PR. Carbamazepine versus phenytoin monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2009, Issue 3, Art. No.: CD001911. DOI: 10.1002/14651858.CD001911.pub2.
53. [http://www.drugbank.ca/drugs/DB00252]. 02/04/2010.
54. Rogawski MA, Basil CW. New Molecular Targets for Antiepileptic Drugs: alfa2delta, SV2A, and Kv7/KCNQ/M Potassium Channels. Curr Neurol Neurosci Rep 2008; 8: 345-52.
55. Fisher RS, Sachdeo RC, Pellock J, Penovich PE, Magnus L, Bernstein P. Rapid initiation of gabapentin: a randomized, controlled trial. Neurology 2001; 56: 743-8.
56. French JA, Kanner AM, Bautista J, Abou-Khalil B, Browne T, Harden CL Theodore WH, Bazil C, Stern J, Schachter SC, Bergen D, Hirtz D, Montouris GD, Nespeca M, Gidal B, Marks WJ Jr, Turk WR, Fischer JH, Bourgeois B, Wilner A, Faught RE Jr, Sachdeo RC, Beydoun A, Glauser TA; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology; Quality Standards Subcommittee of the American Academy of Neurology; American Epilepsy Society. Efficacy and tolerability of the new antiepileptic drugs II: treatment of refractory epilepsy: report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2004; 62: 1261-73.
57. Chadwick DW, Anhut H, Greiner MJ, Murray JH, Garofalo EA, Pierce MW, and the International Gabapentin Monotherapy Study Group. A double blind trial of gabapentin monotherapy for newly diagnosed parcial seizures. Neurology 1998; 51: 1282-8.
58. Beydoun A. Monotherapy trials with gabapentin for partial epilepsy. Epilepsia 1999; 40 (suppl 6): S13-6; S73-4.
59. Appleton R, Fichtner K, LaMoreaux L, Alexander J, Halsall G, Murray G, Garofalo E, and the Gabapentin Pedriatric Study Group. Gabapentin as add-on therapy in children with refractory parcial seizures. Epilepsia 1999; 40: 1147-54.
60. Gidal BE, DeCerce J, Bockbrader HN, Gonzalez J, Kruger S, Pitterle ME, Rutecki P, Ramsay RE. Gabapentin bioavailability: effect of dose and frequency of administration in adult patients with epilepsy. Epilepsy Res 1998; 31: 91-9.
61. Patsalos PN, Perucca E. Clinically important drug interactions in epilepsy: general features and interactions between antiepileptic drugs. Lancet Neurol 2003; 2: 347-56.
62. Patsalos PN, Perucca E. Clinically important drug interactions in epilepsy: interactions between antiepileptic drugs and other drugs. Lancet Neurol 2003; 2: 473-81.
63. Pellock JM, Appleton R. Use of new antiepileptic drugs in the treatment of childhood epilepsy. Epilepsia 1999; 40 (suppl 6): S29-S38.
64. Bourgeois B. New dosages and formulations of AEDs for use in pediatric epilepsy. Neurology 2002; 58 (Suppl 7): S2-S5.
65. Loring DW, Meador KJ. Cognitive and behavioral effects of epilepsy treatment. Epilepsia 2001; 42 (Suppl 8): 24.
66. Montouris G. Gabapentin exposure in human pregnancy: results from the Gabapentin Pregnancy Registry. Epilepsy Behav 2003; 4: 310-7.
67. Marson AG, Kadir ZZ, Hutton JL, Chadwick DW. Gabapentin add-on for drug-resistant partial epilepsy. Cochrane Database of Systematic Reviews 1999, Issue 1. Art. No.: CD001415. DOI: 10.1002/14651858.CD001415.
68. Mattson RH, Cramer JA, Collins JF, et al. Comparision of carbamazepine, Phenobarbital, phenytoin, and primidone in partial and secondarily generalized tonic-clonic seizures. N Eng J Med 1985;313: 145-51.
69. Murphy K, Delanty N. Primary generalized epilepsies. Curr Treat Options Neurol 2000; 2(6): 527-42.9
70. Jette N, Hemming K, Hutton JL, Marson AG. Topi-ramate add-on for drug-resistant partial epilepsy. Cochrane Database of Systematic Reviews 2008, Issue 3. Art. No.: CD001417. DOI: 10.1002/14651858.CD001417.pub2.
71. Ben-Menachem E, Sander JW, Stefan H, Schwalen S, Schäuble B. Topiramate monotherapy in the treatment of newly or recently diagnosed epilepsy. Clin Ther 2008; 30: 1180-95.
72. Gilliam FG, Veloso F, Bomhof MA, Gazda SK, Biton V, Ter Bruggen JP, Neto W, Bailey C, Pledger G, Wu SC. A dosecomparision trial of topiramate as monotherapy in recently diagnosed partial epilepsy. Neurology 2003; 60: 196-202.
73. Arroyo S, Dodson WE, Privitera MD, Glauser TA, Naritoku DK, Dlugos DJ, Wang S, Schwabe SK, Twyman RE; EPMN106/INT-28 Investigators. Randomized dose-controlled study of topiramate as first-line therapy in epilepsy. Acta Neurol Scand 2005;112: 214-22.
74. Privitera MD, Brodie MJ, Mattson RH, Chadwick DW, Neto W, Wang S; EPMN 105 Study Group. Topiramate, carbamazepine and valproate monotherapy: double-blind comparison in newly diagnosed epilepsy. Acta Neurol Scand 2003; 107: 165-75.
75. Glauser TA, Dlugos DJ, Dodson WE, Grinspan A, Wang S, Wu SC; EPMN-106/INT-28 Investigators. Topiramate monotherapy in newly diagnosed epilepsy in children and adolescents. J Child Neurol 2007; 22: 693-9.
76. de Araujo Filho GM, Pascalicchio TF, Lin K, Sousa PS, Yacubian EM. Neuropsychiatric profiles of patients with juvenile myoclonic epilepsy treated with valproate or topiramate. Epilepsy Behav 2006; 8: 606-9.
77. Hancock EC, Cross HH. Treatment of Lennox-Gastaut syndrome. Cochrane Database Syst Rev 2009 Jul 8; (3): CD003277.
78. Landmark CJ. Targets for antiepileptic drugs in the synapse. Med Sci Monit, 2007; 13: 1-7.
79. Saetre E, Perucca E, Isojärvi J, Gjerstad L; LAM 40089 Study Group. An international multicenter randomized double-blind controlled trial of lamotrigine and sustained-release carbamazepine in the treatment of newly diagnosed epilepsy in the elderly. Epilepsia 2007; 48: 1292-302.
80. Sabers A, Tomson T. Managing antiepileptic drugs during pregnancy and lactation. Curr Opin Neurol 2009; 22: 157-61.
81. Sabers A, Petrenaite V. Seizure frequency in pregnant women treated with lamotrigine monotherapy. Epilepsia 2009; 5: 2163-6.
82. Montouris G, Abou-Khalil B. The first line of therapy in a girl with juvenile myoclonic epilepsy: should it be valproate or a new agent? Epilepsia 2009; 50 (Suppl 8): 16-20.
83. Sazgar M, Bourgeois BF. Aggravation of epilepsy by antiepileptic drugs. Pediatr Neurol 2005; 33: 227-34.
84. Ramaratnam S, Marson AG, Baker GA. Lamotrigine add-on for drug-resistant partial epilepsy. Cochrane Database of Systematic Reviews 2001, Issue 3. Art. No.: CD001909. DOI: 10.1002/14651858.CD001909.
85. Willmore LJ, Abelson MB, Ben-Menachem E, Pellock JM, Shields WD. Vigabatrin: 2008 update. Epilepsia 2009; 50: 163-73.
86. Banin E, Shalev RS, Obolensky A, Neis R, Chowers I, Gross-Tsur V. Retinal function abnormalities in patients treated with vigabatrin. Arch Ophthalmol 2003; 121: 811-6.
87. Kalviainen R, Aikia M, Saukkonen AM, Mervaala E, Riekkinen PJ Sr. Vigabatrin vs carbamazepine monotherapy in patients with newly diagnosed epilepsy. A randomized, controlled study. Arch Neurol 1995; 52: 989-96.
88. Marciani MG, Maschio M, Spanedda F, Iani C, Gigli GL, Bernardi G. Development of myoclonus in patients with partial epilepsy during treatment with vigabatrin: an electroencephalographic study. Acta Neurol Scand 1995; 91: 1-5.
89. Hancock EC, Osborne JP, Edwards SW. Treatment of infantile spasms. Cochrane Database Syst Rev 2008 Oct 8;(4):CD001770.
90. Hemming K, Maguire MJ, Hutton JL, Marson AG. Vigabatrin for refractory partial epilepsy. Cochrane Database Syst Rev 2008 Jul 16;(3):CD007302.
91. Tanganelli P, Regesta G. Vigabatrin vs. carbamazepine monotherapy in newly diagnosed focal epilepsy: a randomized response conditional cross-over study. Epilepsy Res 1996; 25: 257-62.
92. Bruni J, Guberman A, Vachon L, Desforges C. Vigabatrin as add-on therapy for adult complex partial seizures: a double-blind, placebo-controlled multicentre study. The Canadian Vigabatrin Study Group. Seizure 2000; 9: 224-32.
93. Chadwick D. Safety and efficacy of vigabatrin and carbamazepine in newly diagnosed epilepsy. Lancet 1999; 354: 13-9.
94. Zamponi N, Cardinali C. Open comparative long-term study of vigabatrin vs carbamazepine in newly diagnosed partial seizures in children. Arch Neurol 1999; 56: 605-7.
95. Marson AG, Williamson PR, Hutton JL, Clough HE, Chadwick DW. Carbamazepine versus valproate monotherapy for epilepsy. Cochrane Database of Systematic Reviews 2000, Issue 3. Art. No.: CD001030. DOI: 10.1002/14651858.CD001030.
96. Smith CT, Marson AG, Chadwick DW, Williamson PR. Multiple treatment comparisons in epilepsy monotherapy trials. Trials 2007; 8: 34.
97. Brodie MJ, Yuen AW. Lamotrigine substitution study: evidence for synergism with sodium valproate? 105 Study Group. Epilepsy Res 1997; 26: 415-32.
98. Pisani F, Oteri G, Russo MF, Di Perri R, Perucca E, Richens A. The efficacy of valproate-lamotrigine comedicaton in refractory complex partial seizures: evidence for a pharmacodynamic interaction. Epilepsia 1999; 40: 1141-6.
99.Rowan AJ, Meijer JWA, de Beer-Pawlikowski N, van der Geest P, Meinardi H. Valproate-ethosuximide combination therapy for refractory abdsence seizures. Arch. Neurol 1983; 40: 797-802.
100. Cereghino JJ, Brock JT, Van-Meter JC, Penry JK, Smith LD, White BG. The efficacy of carbamazepine combination in epilepsy. Clin Pharmacol Ther 1975; 18: 733-41.
101. Brodie MJ, Mumford JP. Double-blind substitution of vigabatrin and valproate in carbamazepine-resistant partial epilepsy. 012 Study Group. Epilepsy Res. 1999; 34: 199-205.
102. Leach JP, Brodie MJ. Synergism with GABA-ergic drugs in refractory epilepsy. Lancet 1994; 343: 1650.
103. Stephen LJ, Sills GJ, Brodie MJ. Lamotrigine and topiramate may be a useful combination. Lancet 1998; 351: 958-9.
104. Wiebe S, Blume WT, Girvin JP, Eliasziw M; Effectiveness and Efficiency of Surgery for Temporal Lobe Epilepsy Study Group. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med. 2001; 345: 311-8.
105. Kwan P, Brodie MJ. Combination therapy in epilepsy: when and what to use. Drugs 2006; 66: 1817-29.
106. Sirven J, Sperling MR, Wingerchuk DM. Early versus late antiepileptic drug withdrawal for people with epilepsy in remission. Cochrane Database of Systematic Reviews 2001, Issue 3. Art. No.: CD001902. DOI: 10.1002/14651858.CD001902.
TERMO DE ESCLARECIMENTO E RESPONSABILIDADE
CLOBAZAM, ETOSSUXIMIDA, GABAPENTINA, PRIMIDONA, TOPIRAMATO, LAMOTRIGINA E VIGABATRINA.
Eu,__________________________(nome do(a) paciente), declaro ter sido informado(a) claramente sobre benefícios, riscos, contraindicações e principais efeitos adversos relacionados ao de CLOBAZAM, ETOSSUXIMIDA, GABAPENTINA, PRIMIDONA, TOPIRAMATO, LAMOTRIGINA E VIGABATRINA, indicados para o tratamento da EPILEPSIA.
Os termos médicos foram explicados e todas as minhas dúvidas foram resolvidas pelo médico ____________________ (nome do médico que prescreve).
Assim, declaro que fui claramente informado(a) de que o medicamento que passo a receber pode trazer as seguintes melhoras:
- diminuição dos eventos convulsivos;
- melhora da qualidade de vida.
Fui também claramente informado(a) a respeito das seguintes contraindicações, potenciais efeitos adversos e riscos do uso do medicamento:
- não se sabe ainda ao certo os riscos do uso de primidona, lamotrigina, gabapentina, topiramato na gravidez; portanto, caso engravide, devo avisar imediatamente o médico;
- clobazam e etossuximida não podem ser usados durante a gravidez pelo risco de má formação do feto;
- vigabatrina apresenta risco na gravidez, porém o beneficio pode ser maior do que o risco; portanto, caso engravide, devo avisar imediatamente o médico;
- efeitos adversos da clobazam -ansiedade, boca seca, coceiras, prisão de ventre, dor de cabeça, cansaço, náuseas, vômitos, perda de memória, sonolência, vermelhidão na pele;
- efeitos adversos da etossuximida - tontura, sonolência, dor de cabeça, soluços, perda de peso, náuseas, vômitos, reações alérgicas, com aparecimentos de lesões de pele potencialmente graves, incluindo a síndrome de Stevens-Johnson, irritabilidade, dificuldade de concentração, pesadelos, alterações nas células do sangue (raramente);
- efeitos adversos da gabapentina -diminuição das células brancas do sangue, constipação, secura na boca, náuseas, vômitos, tontura, sonolência, cansaço, depressão, confusão, nervosismo, descoordenação, amnésia, ganho de peso, visão turva ou dupla, coceira na pele, rinite, bronquite, faringite, tosse e infecções respiratórias, edema periférico, febre;
- efeitos adversos da primidona -tonturas, sonolência, problemas de coordenação motora, problemas na pele, dor nas juntas, febre, problemas gastrointestinais, náuseas, vômitos, perda de apetite, problemas nos olhos;
- efeitos adversos da topiramato - náuseas, dores abdominais, tonturas, fadiga, sonolência, cansaço, dificuldade de concentração ou atenção, nervosismo, irritabilidade, agressão, agitação, dificuldade de expressão verbal, confusão, depressão, edema, diminuição da audição, problemas para urinar, sangue na urina, febre, perda de apetite, perda de peso, cefaleia, coceiras, diminuição do apetite sexual ou impotência, alterações no ciclo menstrual, conjuntivite;
- efeitos adversos da lamotrigina -reações alérgicas, com aparecimentos de lesões de pele potencialmente graves (síndrome de Stevens-Johnson), estando relacionadas com aumento abrupto da dose, diminuição das células brancas, vermelhas e plaquetas do sangue, constipação ou diarreia, secura na boca, indigestão, náuseas, vômitos, dor abdominal, inflamação no esôfago, inflamação no pâncreas, tontura, sonolência, dor de cabeça, irritabilidade, depressão, descoordenação, tremores, amnésia, perda de peso, visão turva ou dupla, alterações no ciclo menstrual, febre;
- efeitos adversos da vigabatrina -diminuição das células vermelhas do sangue, constipação, secura na boca, náuseas, vômitos, dor de estômago, tontura, dor de cabeça, depressão, confusão, nervosismo, dificuldade de concentração, sonolência, cansaço, ganho de peso, crescimento da gengivas, visão dupla, reações alérgicas de pele;
- contraindicado em casos de hipersensibilidade (alergia) aos componentes da fórmula;
- risco da ocorrência de efeitos adversos aumenta com a superdosagem e com o uso concomitante de outros medicamentos.
Estou ciente de que este medicamento somente pode ser utilizado por mim, comprometendo-me a devolvê-lo caso não queira ou não possa utilizá-lo ou se o tratamento for interrompido. Sei também que continuarei a ser atendido(a), inclusive em caso de desistir de usar o medicamento.
Autorizo o Ministério da Saúde e as Secretarias de Saúde a fazerem uso de informações relativas ao meu tratamento, desde que assegurado o anonimato.
O meu tratamento constará do(s) seguinte(s) medicamento(s):
( ) clobazam
( ) etossuximida
( ) gabapentina
( ) primidona
( ) topiramato
( ) lamotrigina
( ) vigabatrina
| Local: Data: | ||
| Nome do paciente: | ||
| Cartão Nacional de Saúde: | ||
| Nome do responsável legal: | ||
| Documento de identificação do responsável legal: | ||
| Assinatura do paciente ou do responsável legal | ||
| Médico responsável: | CRM: | UF: |
| Assinatura e carimbo do médico Data:____________________ | ||
OBSERVAÇÃO: Este Termo é obrigatório para solicitação de medicamento do Componente Especializado da Assistência Farmacêutica e deverá ser preenchido em duas vias: uma será arquivada na farmácia, e a outra, entregue ao usuário ou a seu responsável legal.
Quadro resumo com as indicações dos medicamentos para cada tipo de peilepsia, baseado no texto deste PCDT