Este texto não substitui o publicado no Diário Oficial da União

Agência Nacional de Vigilância Sanitária
RESOLUÇÃO-RDC Nº 199, DE 12 DE JULHO DE 2002
A Diretoria Colegiada da Agência Nacional de Vigilância Sanitária no uso da atribuição que lhe confere inciso IV do art. 13 do Regulamento da ANVISA aprovado pelo Decreto nº 3.029, de 16 de abril de 1999, em reunião realizada em 10 de julho de 2002.
considerando o inciso XIX do art. 7º da Lei nº 9.782, de 26 de janeiro de 1999;
adotou a seguinte Resolução e eu, Diretor-Presidente, determino a sua publicação:
Art. 1º Fica aprovado o Fascículo 3 da Parte II da 4ª Edição da Farmacopéia Brasileira, em anexo, elaborado pela Comissão Permanente de Revisão da Farmacopéia Brasileira-CPRFB, instituída pela Portaria nº 12-ANVS, de 20 de janeiro de 2000.
Art. 2º Esta Resolução de Diretoria Colegiada entra em vigor na data de sua publicação.
GONZALO VECINA NETO
PARTE II
A identificação das monografias na Parte II é efetuada pelo número de série e o ano da publicação de sua última versão. Os textos da Parte I são identificados pelo número de referência e o ano de publicação da última versão.
Os textos e monografias publicados no presente Fascículo anulam os textos e monografias publicados, anteriormente, nesta edição ou em outras edições da Farmacopéia Brasileira.
III COMISSÃO PERMANENTE DE REVISÃO DA FARMACOPÉIA BRASILEIRA
MINISTÉRIO DE ESTADO DA SAÚDE
BARJAS NEGRI
AGÊNCIA NACIONAL DE VIGILÂNCIA SANITÁRIA
DIRETOR- PRESIDENTE
GONZALO VECINA NETO
DIRETORIA COLEGIADA
GONZALO VECINA NETO
CLÁUDIO MAIEROVITCH PESSANHA HENRIQUES
LUIS CARLOS WANDERLEY LIMA
LUIZ MILTON VELOSO COSTA
RICARDO OLIVA
COMISSÃO PERMANENTE DE REVISÃO
DA FARMACOPÉIA BRASILEIRA
PRESIDENTE
CELSO F. BITTENCOURT
CYPRIANO CARDOSO FILHO
Farmacêutico
Associação Brasileira de Farmacêuticos
Rio de Janeiro, RJ
EDUARDO AUGUSTO MOREIRA
Professor
Curso de Farmácia da Universidade Federal
do Paraná
Curitiba, PR
EDUARDO CHAVES LEAL
Farmacêutico
Instituto Nacional de Controle de Qualidade
em Saúde/FIOCRUZ
Rio de Janeiro, RJ
ELFRIDES E. SCHERMAN SCHAPOVAL
Professora
Faculdade de Farmácia da Universidade Federal do Rio Grande do
Sul
Porto Alegre, RS
ELIZABETH IGNE FERREIRA
Professora
Faculdade de Ciências Farmacêuticas
da Universidade de São Paulo
São Paulo, SP
ELZA ANDERS SAAD
Farmacêutica
União Farmacêutica de São Paulo
São Paulo, SP
GERALDO FENERICH
Farmacêutico
Agência Nacional de Vigilância Sanitária
do Ministério da Saúde
Brasília, DF
GERSON ANTÔNIO PIANETTI
Professor
Faculdade de Farmácia da Universidade Federal de Minas Gerais
Belo Horizonte, MG
JOÃO CARLOS PALAZZO DE MELLO
Farmacêutico
Conselho Federal de Farmácia
Brasília, DF
LAURO DOMINGOS MORETTO
Farmacêutico
Sindicato da Indústria de Produtos Farmacêuticos
no Estado de São Paulo
São Paulo, SP
NIKOLAI SHARAPIN
Professor
Faculdade de Farmácia da Universidade Federal Fluminense
Niterói, RJ
SALVADOR ALVES PEREIRA
Professor
Faculdade de Farmácia da Universidade Federal Fluminense
Niterói, RJ
SUBCOMISSÕES DA COMISSÃO PERMANENTE DE REVISÃO
DA FARMACOPÉIA BRASILEIRA
SUBCOMISSÃO DE CORRELATOS
Dhalia Gutemberg
Therezinha de Jesus Andreoli Pinto
Márcia Aparecida Aguiar
Isabel Kendall
SUBCOMISSÃO DE DENOMINAÇÕES COMUNS BRASILEIRAS
Aulus Conrado Basile
Fátima Goulart Farhat
Elizabeth Igne Ferreira
Raquel Ribeiro Bittencourt
Carlos Vidoti
SUBCOMISSÃO DE EXCIPIENTES E ADJUVANTES
José Aparício Brittes Funck
Mauro Witzel
Marcos Paulo Moreira
Ana Maria Braguim Pellim
Armando da Silva Cunha
Valéria Cozzolivo Yugue
SUBCOMISSÃO DE FITOTERÁPICOS
Eduardo Augusto Moreira
Nikolai Sharapin
Leandro Machado Rocha
Célia Helena Ognibene
Melânia Palermo Manfron
Luiz Antônio da Costa
Elfriede Marianne Bacchi
SUBCOMISSÃO DO FORMULÁRIO NACIONAL
Salvador Alves Pereira
David Telvio Knobel
Elpidio Nereu Zanchet
Julio Fernades Maia Neto
Luiz Fernando Chiavegatto
Marco Antônio Perino
Paulo Queiroz Marques
Rogério Tokarski
Victor Hugo Travassos da Rosa
Celso F. Bittencourt
Nikolai Sharapin
Alexandre Fiuza Juliano
Luciane Varini Laporta
SUBCOMISSÃO DE HOMEOPATIA
Gilberto Luiz Pozetti
Edanir dos Santos
Elza Helena Guimarães Lara
Luiz Cezar de Camargo Carvalho
Marília Bortoluzzi
Maria Izabel Almeida Prado
Renan Ruiz
Margareth de Akemi Kishi
SUBCOMISSÃO DE IMUNOBIOLÓGICOS
Eduardo Chaves Leal
Darcy Akemi Hokama
João Carlos Repka
Hisako Higashi
Lilia Ribeiro Seródio
Kleide de Carvalho Teixeira
Maria Irene G. Narciso
Carlos Nozawa
SUBCOMISSÃO DE MATERIAL DE REFERÊNCIA
André Luiz Gemal
Celso F. Bittencourt
Augusto Bortoluzzi
Pedro Eduardo Fröhelich
Lauro Domingos Moretto
Érico Marlon Flores
Sérgio Luiz Dalmora
Maria Inês M. Santoro
Maria do Carmos Vasques Garcia
SUBCOMISSÃO DE PLANTAS MEDICINAIS
Amélia T. Henriques
Elfriede Marianne Bacchi
José Ângelo Zuanazzi
Paulo Luiz de Oliveira
Lílian Auler Mentz
Leandro Machado Rocha
Eduardo Augusto Moreira
Nikolai Sharapin
João Carlos Palazzo de Mello
José Luiz Pinto Ferreira
SUBCOMISSÃO DE REVISÃO E HARMONIZAÇÃO
Ligia Maria Moreira de Campos
Antônio Basílio Pereira
Nilton de Souza Junior
Maria Auxiliadora Prado
SUBCOMISSÃO DE SUBSTÂCIAS BIOLÓGICAS
Sérgio Luiz Dalmora
Maria Virginea Scarpa Oliveira
Paolo Bartolini
Célia Gervásio Chaves
Marco Aurélio Xavier
Mitsuko Taba Ohara
Octavio França Presgrave
COLABORADORES DO FASCÍCULO 3
AKIMI MORI HONDA
Farmacêutica
Assessora da Diretoria Industrial e Professora
Farmasa - Laboratório Americano de Farmacoterapia S/A e Curso de
Farmácia da UNIABC
São Paulo, SP
ALCIDES GUIMARÃES DA ROCHA
Químico Industrial
Gerente Controle de Qualidade da
Pharmacia Brasil Ltda
São Paulo, SP
ALCIDES HORIE
Farmacêutico
Gerente Controle de Qualidade
FURP - Fundação Para o Remédio Popular
Guarulhos, SP
AMÉLIA T. HENRIQUES
Professora
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
ANA CRISTINA R. CORREIA
Farmacêutica
Departamento de Ciências Farmacêuticas
Universidade Federal de Pernambuco
Recife, PE
ANA FLÁVIA OLIVEIRA SANTOS
Bolsista da CPRFB
Faculdade de Farmácia da
Universidade Federal da Paraíba
João Pessoa, PB
ANA LAURA VENQUIARUTI ESCARRONE
Bolsista da CPRFB
Curso de Farmácia e Bioquímica
Universidade Federal de Santa Maria
Santa Maria, RS
ANA LÚCIA BOY
Bolsista da CPRFB
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
ANA PAULA PEREIRA BRITO
Farmacêutica
Faculdade de Farmácia da
Universidade Federal do Rio de Janeiro
Rio de Janeiro, RJ
ANDRÉ LUIZ GEMAL
Diretor
Instituto Nacional de Controle de Qualidade em Saúde/FIOCRUZ
Rio de Janeiro, RJ
ANGÉLICA GARCIA COUTO
Farmacêutica
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
ANTÔNIA DE ARAÚJO OLIVEIRA
Farmacêutica
Gerente de Controle de Qualidade
Aventis Pharma Ltda
São Paulo, SP
ANTÔNIO BASÍLIO PEREIRA
Professor
Faculdade de Farmácia
Universidade Federal de Minas Gerais
Belo Horizonte, MG
ANTONIO FERNANDO RIBEIRO DA SILVA
Farmacêutico
Microbiológica e Química Farmacêutica Ltda
Rio de Janeiro, RJ
AUGUSTO VILSON BORTOLUZZI
Professor
Curso de Farmácia e Bioquímica da
Universidade Federal da Santa Maria
Santa Maria, RS
BRENO DE CARVALHO E SILVA
Bolsista da CPRFB
Faculdade de Farmácia
Universidade Federal de Minas Gerais
Belo Horizonte, MG
CAMILA FRANCO
Bolsista da CPRFB
Curso de Ciências Farmacêuticas do
Centro Universitário Franciscano
Santa Maria, RS
CARLOS EDUARDO B. LIMA
Químico
Chefe de Seção Matéria-Prima/Material de Embalagem
Novartis Biociências S.A
Taboão da Serra, SP
CÁSSIA VIRGINIA GARCIA
Bolsista da CPRFB
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
CARLOS EDUARDO B. LIMA
Químico
Chefe de Seção Matéria-Prima/Material de Embalagem
Novartis Biociências S.A
Taboão da Serra, SP
CÉLIA DE FREITAS GUIMARÃES PRAÇA
Professora
Faculdade de Farmácia da
Universidade Federal do Ceará
Fortaleza, CE
CÉLIA YOKO SASAKI
Farmacêutica
Assessora de Qualidade da
União Química Farmacêutica S.A e
Biolab Sanus Farmacêutica Ltda
Taboão da Serra, SP
CELINA YUMI MOTIZUKI
Bolsista da CPRFB
Faculdade de Ciências Farmacêuticas da
Universidade de São Paulo
São Paulo, SP
CELSO F. BITTENCOURT
Professor
Curso de Farmácia e Bioquímica da
Universidade Federal de Santa Maria
Santa Maria, RS
CHRISTIAN FERNANDES
Farmacêutico
Faculdade de Farmácia da
Universidade Federal de Minas Gerais
Belo Horizonte, MG
CINTIA SATIE QUICU
Química
Aventis Pharma Ltda
Suzano, SP
CLAUDIO VALÉRIO BORTALIERO
Farmacêutico
Supervisor de CTC&QC do
Laboratório Stiefel Ltda
Guarulhos, SP
CLÉSIO SOLDATELLI PAIM
Bolsista da CPRFB
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
CLEYTON EDUARDO M. DE TOLEDO
Bolsista da CPRFB
Curso de Farmácia da
Universidade Estadual de Maringá
Maringá, PR
CRISTIANE FRANCO CODEVILLA
Farmacêutica
Curso de Farmácia e Bioquímica da
Universidade Federal de Santa Maria
Santa Maria, RS
CYPRIANO CARDOSO FILHO
Farmacêutico
Associação Brasileira de Farmacêuticos
Rio de Janeiro, RJ
DANIEL HENRIQUES SOARES LEAL
Bolsista da CPRFB
Faculdade de Farmácia da
Universidade Federal de Minas Gerais
Belo Horizonte, MG
DANIELLE COUTINHO LORDÃO
Bolsista da CPRFB
Departamento de Ciências Farmacêuticas da
Universidade Federal de Pernambuco
Recife, PE
DANTE ALARIO JUNIOR
Farmacêutico
Diretor Técnico da
Biolab Sanus Farmacêutica Ltda
São Paulo, SP
DÁRCIO CALLIGARIS
Farmacêutico
Fundação para o Remédio Popular/FURP
São Paulo, SP
DÉBORA GLITZENHIRN
Bolsista da CPRFB
Curso de Farmácia e Bioquímica
Universidade Federal de Santa Maria
Santa Maria, RS
DENIZE CÁSSIA RESENDE
Farmacêutica
Faculdade de Farmácia
Universidade Federal de Minas Gerais
Belo Horizonte, MG
DENISE DAVANÇO PELEGRINI
Bolsista da CPRFB
Curso de Farmácia da
Universidade Estadual de Maringá
Maringá, PR
DILZA VARANDAS
Farmacêutica
Gerente de Controle de Qualidade
Sanofi-Synthelabo Ltda
São Paulo, SP
EDUARDO ALMEIDA GOMES
Bolsista da CPRFB
Fundação Oswaldo Cruz
Rio de Janeiro, RJ
EDUARDO AUGUSTO MOREIRA
Professor
Curso de Farmácia da
Universidade Federal do Paraná
Curitiba, PR
EDUARDO CHAVES LEAL
Farmacêutico
Instituto Nacional de Controle de Qualidade
em Saúde/FIOCRUZ
Rio de Janeiro, RJ
ELAINE C. M. PESSOA
Farmacêutica
Gerente de Controle de Qualidade
Bunker Industria Farmacêutica Ltda
São Paulo, SP
ELAINE DE FERITAS MAGATONI
Química Industrial
Supervisora de Controle de Qualidade
Asta Médica Ltda
São Paulo, SP
ELFRIDES E. SCHERMAN SCHAPOVAL
Professora
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
ELIANA C. M. NUNES
Professora
Instituto de Biociências da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
ELIANE SOUZA CARVALHO
Professora
Faculdade de Farmácia da
Universidade Federal Fluminense
Niterói, RJ
ELIZABETH DE ALBUQUERQUE LÚCIO
Bolsista da CPRFB
Instituto de Química da
Universidade Federal Fluminense
Niterói, RJ
ELIZABETH IGNE FERREIRA
Professora
Faculdade de Ciências Farmacêuticas da
Universidade de São Paulo
São Paulo, SP
ELZA ANDERS SAAD
Farmacêutica
União Farmacêutica de São Paulo
São Paulo, SP
ÉRICO MARLON DE MORAES FLORES
Professor
Curso de Química Industrial da
Universidade Federal de Santa Maria
Santa Maria, RS
ÉRIKA M. MATSUMOTO
Farmacêutica
Indústria Química e Farmacêutica Schering-Plough S/A
Jacarepaguá, RJ
FABIANE MOREIRA FARIAS
Farmacêutica,
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
FÁBIO SANTOS DE SOUZA
Farmacêutico
Faculdade de Farmácia da
Universidade Federal da Paraíba
João Pessoa, PB
FERNANDA POLETTO
Bolsista da CPRFB
Faculdade de Farmácia da
Universidade de Passo Fundo
Passo Fundo, RS
FERNANDA RUNHA
Farmacêutica
Gerente de Garantia de Qualidade
Jansen-Cilag Farmacêutica Ltda
São Paulo, SP
FLÁVIA MARIANO PINTO
Técnica Química
Analista Júnior de Laboratório
Asta Médica Ltda
São Paulo, SP
FLAVIA PEREIRA ADÃO
Farmacêutica
Analista Controle de Qualidade
Indústria Química e Farmacêutica Schering-Plough S/A
Jacarepaguá, RJ
FLAVIO VALENTE
Farmacêutico
Gerente de Produtos
Nortec Química Desenvolvimento Tecnológico
São Paulo, SP
GERALDO FENERICH
Farmacêutico
Agência Nacional de Vigilância Sanitária
Ministério da Saúde
Brasília, DF
GERSON ANTÔNIO PIANETTI
Professor
Faculdade de Farmácia da
Universidade Federal de Minas Gerais
Belo Horizonte, MG
GIZELE SILVA CRUVINEL
Bióloga
Supervisora da Microbiologia
Laboratórios Pfizer Ltda
Guarulhos, SP
H. J. KILLIAN
Farmacêutico
Diretor Industrial
BYK Química Farmacêutica Ltda
Jaguariúna, SP
HELCIO LA SCALA TEIXEIRA
Farmacêutico
Diretor de Qualidade
Jansen-Cilag Farmacêutica Ltda
São Paulo, SP
IVETE BORTOLUCCI
Química
Gerente Garantia da Qualidade
BYK Química Farmacêutica Ltda
Jaguariúna, SP
IVONE SARTOR
Professora
Curso de Farmácia da
Universidade Católica do Rio Grande do Sul
Porto Alegre, RS
JANE BEATRIZ LIMBERGER
Bolsista da CPRFB
Curso de Farmácia e Bioquímica
Universidade Federal de Santa Maria
Santa Maria, RS
JEANNE DE ARAÚJO C. PEREIRA
Química
Instituto Nacional de Controle de Qualidade em Saúde/ FIOCRUZ
Rio de Janeiro, RJ
JOÃO CARLOS PALAZZO DE MELLO
Professor
Curso de Farmácia da
Universidade Estadual de Maringá
Maringá, PR
JORGE COSTA
Farmacêutico
Gerente de Controle de Qualidade
Nortec Química Desenvolvimento Tecnológico
São Paulo, SP
JOSÉ ÂNGELO SILVEIRA ZUANAZZI
Professor
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
JOSÉ APARÍCIO BRITTES FUNCK
Professor
Curso de Ciências Farmacêuticas do
Centro Universitário Franciscano
Santa Maria, RS
JOSÉ LUIS PINTO FERREIRA
Professor
Faculdade de Farmácia da
Universidade Federal Fluminense
Niterói, RJ
JOSÉ MARIA LOPES DE ALMEIDA
Professor
Faculdade de Farmácia da
Universidade Federal Fluminense
Niterói, RJ
JOSÉ ROBERTO F. DE ALMEIDA
Químico
Superintendente Industrial
Laboratório Sintofarma S/A
Tabuão da Serra, SP
JULIANO SMANIOTO BARIN
Farmacêutico
Curso de Química Industrial da
Universidade Federal de Santa Maria
Santa Maria, RS
JULIO CÉSAR CAJARANA
Farmacêutico Bioquímico
Pesquisador
E.M.S. Industria Farmacêutica Ltda
São Paulo, SP
JULIO CESAR CARESTIATO
Professor
Faculdade de Farmácia da
Universidade Federal Fluminense
Niterói, RJ
KELLEN CRISTHINA BORGES DE SOUZA
Farmacéutica
Faculdade de Farmácia da
Universidade Fedreral do Rio Grande do Sul
Porto Alegre, RS
KELLY CHRISTINE DA SILVA CARNEIRO
Farmacêutica
Departamento de Ciências Farmacêuticas da
Universidade Federal de Pernambuco
Recife, PE
LAURO DOMINGOS MORETTO
Farmacêutico
Sindicato da Indústria de Produtos Farmacêuticos no Estado de São
Paulo
São Paulo, SP
LÁZARO DE JESUS GAMBARELI
Farmacêutico
Gerente de Garantia e Controle de Qualidade
ICN Farmacêutica Ltda
Campinas, SP
LEANDRO MACHADO ROCHA
Professor
Faculdade de Farmácia da
Universidade Federal Fluminense
Niterói, RJ
LENISE ARNEIRO TEIXEIRA
Professora
Faculdade de Farmácia da
Universidade Federal Fluminense
Niterói, RJ
LÍGIA CRISTINA DIRESZ
Bolsista da CPRFB
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
LÍGIA MARIA MOREIRA DE CAMPOS
Professora
Faculdade de Farmácia da
Universidade Federal de Minas Gerais
Belo Horizonte, MG
LILIAN AULER MENTZ
Professora
Instituto de Biociências da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
LISIANE BAJERSKI
Farmacêutica
Curso de Farmácia Industrial
Universidade Federal da Santa Maria
Santa Maria, RS
LÚCIA LAGO HAMMES
Farmacêutica
Gerente Garantia de Qualidade
Indústria Química e Farmacêutica Schering-Plough S/A
Jacarepaguá, RJ
LUCIANA OLIVEIRA DOS SANTOS
Química
Instituto Nacional de Controle de Qualidade em Saúde / FIOCRUZ
Rio de Janeiro, RJ
LUCIANE VARINI LAPORTA
Farmacêutica
Secretária-executiva da CPRFB,
Centro Universitário Franciscano
Santa Maria, RS
LUCILIA CRISTINA SATOMI
Farmacêutica
Faculdade de Ciências Farmacêuticas da
Universidade de São Paulo
São Paulo, SP
LUIS FELIPE DIAS LOPES
Professor
Departamento de Estatística da
Universidade Federal de Santa Maria
Santa Maria, RS
MAGALI BENJAMIN DE ARAÚJO
Professora
Escola de Farmácia e Odontologia
de Alfenas
Alfenas, MG
MAGDA CASSIA CORIOLANO SILVEIRA
Farmacêutica
Gerente de Controle de Qualidade
Astra zeneca Ltda
Cotia, SP
MAGDA TARGA MARTINS
Bolsista da CPRFB
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
MARCELO PTAZSNIK
Químico
Analista Químico Senior
Novartis Biociências S.A
Taboão da Serra, SP
MÁRCIA VIGNOLI DA SILVA
Bolsista da CPRFB
Faculdade de Farmácia
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
MÁRCIO FERRARINI
Farmacêutico
Faculdade de Ciências Farmacêuticas da
Universidade de São Paulo
São Paulo, SP
MARCILIA PINHEIRO DA COSTA
Farmacêutica
Faculdade de Farmácia da
Universidade Federal do Ceará
Fortaleza, CE
MARCO ANTÔNIO SIQUEIRA
Farmacêutico
Diretor de Garantia Qualidade
Novartis Biociência S/A
Taboão da Serra, SP
MARGARIDA TERUKO KATO
Farmacêutica
Chefe do Controle de Qualidade
FURP - Fundação Para o Remédio Popular
Guarulhos, SP
MARIA AMÉLIA BARATA DA SILVEIRA
Professora
Faculdade de Ciências Farmacêuticas da
Universidade de São Paulo
São Paulo, SP
MARIA AUXILIADORA FONTES PRADO
Professora
Faculdade de Farmácia
Universidade Federal de Minas Gerais
Belo Horizonte, MG
MARIA DO CARMO VASQUES GARCIA
Química
Coord. Programa Materiais de Referência
Instituto Nacional de Controle de Qualidade em Saúde / FIOCRUZ
Rio de Janeiro, RJ
MARIA GABRIELA DE ARAÚJO
Bolsista da CPRFB
Faculdade de Farmácia da
Universidade de São Paulo
São Paulo, SP
MARIA INÊS ROCHA MIRITELLO SANTORO
Professora
Faculdade de Ciências Farmacêuticas da
Universidade de São Paulo
São Paulo, SP
MARGARETH LINDE ATHAYDE
Professora
Curso de Farmácia e Bioquímica da
Universidade Federal de Santa Maria
Santa Maria, RS
MARINÊS JOST E SOUZA
Farmacêutica
Curso de Farmácia e Bioquímica da
Universidade Federal de Santa Maria
Santa Maria, RS
MARTHA ANA GATTUSO
Professora
Faculdade Ciências Bioquímicas e Farmacêuticas da
Universidade Nacional de Rosário
Rosário, Argentina
MEIRE FUSHIMI
Farmacêutica
Diretora do Rd Inrl
Laboratórios Stiefel Ltda
Guarulhos, SP
MICHELE SOARES BITTENCOURT
Bolsista da CPRFB
Escola de Farmácia e Bioquímica da
Universidade Católica de Pelotas
Pelotas, RS
MIRACY MUNIZ DE ALBUQUERQUE
Professora
Departamento de Ciências Farmacêuticas da
Universidade Federal de Pernambuco
Recife, PE
NADIA ARACI BOU CHACRA
Farmacêutica
Faculdade de Ciências Farmacêuticas
Universidade São Paulo
São Paulo, SP
NADIA MARIA VOLPATO
Professora
Faculdade de Farmácia da
Universidade Federal do Rio de Janeiro
Rio de Janeiro, RJ
NELSON DE OLIVEIRA
Químico
Auditor
Laboratórios Pfizer Ltda
Guarulhos, SP
NIKOLAI SHARAPIN
Professor
Faculdade de Farmácia da
Universidade Federal Fluminense
Niterói, RJ
NILTON DE SOUZA VIANA JÚNIOR
Farmacêutico
Faculdade de Farmácia
Universidade Federal de Minas Gerais
Belo Horizonte, MG
NILZETE PAIVA DE SOUZA
Química
Instituto Nacional de Controle de Qualidade em Saúde / FIOCRUZ
Rio de Janeiro, RJ
PAULA CRISTINA MADALOZZO
Bolsista da CPRFB
Faculdade de Farmácia da
Universidade Regional Integrada do
Alto Uruguai e das Missões
Erechim, RS
PAULA GIORGI
Farmacêutica
Analista Júnior
Laboratórios Stiefel Ltda.
Guarulhos, SP
PAULO LUIZ DE OLIVEIRA
Professor
Instituto de Biociências da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
PEDRO EDUARDO FROEHLICH
Professor
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
RAQUEL DUARTE DE TOLEDO
Secretária
Sindicato da Indústria de
Produtos Farmacêuticos no Estado de São Paulo
São Paulo, SP
RENATA LOPES DE OLIVEIRA
Farmacêutica
Supervisora Controle de Qualidade
Indústria Química e Farmacêutica Schering-Plough S/A
Jacarepaguá, RJ
RENATA PEREIRA LIMBERGER
Farmacêutica,
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
RENATO MEDEIROS SILVA
Químico
Supervisor do Laboratório de Equivalência
E.M.S. Industria Farmacêutica Ltda.
São Paulo, SP
RICARDO CHIAPPA
Farmacêutico
Secretário da CPRFB
Universidade Federal de Santa Maria
Santa Maria, RS
ROBERTA VINHAS BERTOLINI
Farmacêutica
Analista de Laboratório
FURP - Fundação Para o Remédio Popular
Guarulhos, SP
ROBERTA UTIDA
Química
Coordenadora Desenvolvimento/Validação
BYK Química Farmacêutica Ltda
Jaguariúna, SP
ROBERTO PARISE FILHO
Farmacêutico
Faculdade de Ciências Farmacêuticas da
Universidade de São Paulo
São Paulo, SP
ROSANA NOCE CARNIEL
Química Industrial
Supervisora do Laboratório Químico
Pharmacia Brasil Ltda
São Paulo, SP
ROSSANA MARIA CARVALHO BRAGA
Bolsista da CPRFB
Departamento de Ciências Farmacêuticas da
Universidade Federal de Pernambuco
Recife, PE
ROSECLER DA ROSA KULMANN
Farmacêutica
Curso de Farmácia e Bioquímica da
Universidade Federal de Santa Maria
Santa Maria, RS
RUBENS VINHA JUNIOR
Farmacêutico
Gerente de Garantia de Qualidade
Jansen-Cilag Farmacêutica Ltda
São Paulo, SP
RUI OLIVEIRA MACÊDO
Professor
Faculdade de Farmácia da
Universidade Federal da Paraíba
João Pessoa, PB
RUTE LEA DA SILVA
Química Industrial
Analista de Laboratório Senior
Pharmacia Brasil Ltda.
São Paulo, SP
RUTH RIESINGER STRATTMANN
Farmacêutica
Departamento de Ciências Farmacêuticas da
Universidade Federal de Pernambuco
Recife, PE
SALVADOR ALVES PEREIRA
Professor
Faculdade de Farmácia da
Universidade Federal Fluminense
Niterói, RJ
SERGIO LUIZ DALMORA
Professor
Curso de Farmácia e Bioquímica da
Universidade Federal da Santa Maria
Santa Maria, RS
SEVERINO GRANJEIRO JÚNIOR
Farmacêutico
Faculdade de Farmácia da
Universidade Federal de Pernambuco
Recife, PE
SHIRLEY BARRIOS
Farmacêutica
Supervisora Controle de Qualidade
Akvonobel Divisão Organol - Organon
São Paulo, SP
SIMONE GONÇALVES CARDOSO
Professora
Curso de Farmácia e Bioquímica da
Universidade Federal de Santa Maria
Santa Maria, RS
SOLANGE TEIXEIRA SOARES SANTOS
Farmacêutica
Gerente do Laboratório de Desenvolvimento Analítico
Laboratórios Stiefel Ltda
Guarulhos, SP
SÔNIA ELISABETE CONSTANTE
Arquivista / Desenho e Plástica
Secretária da SCMR da CPRFB
Universidade Federal de Santa Maria
Santa Maria, RS
SUSANA J. GATTUSO
Professora
Faculdade Ciências Bioquímicas e Farmacêuticas da
Universidade Nacional de Rosário
Rosário, Argentina
SUZANA NOGUEIRA
Farmacêutica
Asta Médica Ltda
São Paulo, SP
TERESINHA DE JESUS ANDREOLLI PINTO
Professora
Faculdade de Ciências Farmacêuticas da
Universidade de São Paulo
São Paulo, SP
TÉRCIO PASCHKE OPPE
Professor
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
TOMOKO IMAGURE
Farmacêutica
Coordenadora do Controle de Qualidade
Aventis Pharma Ltda.
Suzano, SP
VALQUIIRIA LINCK BASSANI
Professora
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
VÂNIA FERREIRA DINIZ
Química
Bolsista da Farmacopéia Brasileira / SCMR
FIOCRUZ / INCQS
Rio de Janeiro, RJ
VÂNIA BORTOLETO SABBAF
Química
Gerente de Controle de Qualidade
Asta Médica Ltda
São Paulo, SP
VANESSA MARIA DOS PASSOS MAIO
Farmacêutica
Faculdade de Farmácia da
Universidade Federal do Rio Grande do Sul
Porto Alegre, RS
VIRGÍNIA SCHIANO
Farmacêutica
Gerente de controle de Qualidade
E.M.S. Industria Farmacêutica Ltda.
São Paulo, SP
VIKTORIA DITADI
Farmacêutica
Gerente de controle de Qualidade
Boehringer Ingelheim do Brasil Química e Farmacêutica Ltda
São Paulo, SP
WALMIR COMPIOTTI
Bolsista da CPRFB
Faculdade de Ciências Farmacêuticas da
Universidade de São Paulo
São Paulo, SP
WALMIR PINTO
Químico
Supervisor do Controle de Qualidade
ICN Farmacêutica Ltda.
Campinas, SP
WELLINGTON XAVIER
Farmacêutico
Analista Farmacêutico
Astra Zeneca Ltda
Cotia, SP
WHOCELY VICTOR DE CASTRO
Farmacêutico,
Faculdade de Farmácia da
Universidade Federal de Minas Gerais
Belo Horizonte, MG
MEMBROS DA CPRFB QUE PARTICIPARAM DA ELABORAÇÃO
DA 4ª EDIÇÃO DA FARMACOPÉIA BRASILEIRA
ANDRÉ LUIZ GEMAL
ANDREJUS KOROLKOVAS
ANGELO JOSÉ COLOMBO
ANTÔNIO JOSÉ ALVES
ELIEZER JESUS DE LACERDA BARREIRO
JOÃO GILVAN ROCHA
JOÃO LUIZ DE SANTIAGO DANTAS QUENTAL
JOSÉ ALEIXO PRATES E SILVA
MARIA GISELA PIROS
MARIA JOSÉ MACHADO
PEDRO ROSS PETROVICK
SEBASTIÃO BAETA HENRIQUES
SÉRGIO HENRIQUES FERREIRA
SUZANA MACHADO DE ÁVILA
THEREZINHA C. BARBOSA TOMASSINI
SECRETÁRIOS DE VIGILÂNCIA SANITÁRIA ENVOLVIDOS
NA PUBLICAÇÃO DA 4ª EDIÇÃO DA FARMACOPÉIA BRASILEIRA
ALBERTO FURTADO RAHDE
ANTÔNIO CARLOS ZANINI
BALDUR OSCAR SCHUBERT
ELISALDO LUIZ DE ARAÚJO CARLINI
FRANCISCO DE ASSIS REIS
GONZALO VECINA NETO
JOÃO BATISTA RISI JÚNIOR
JOÃO GERALDO MARTINELLI
JOSÉ ALBERTO HERMÓGENES
JOSÉ RIBEIRO
LUIZ FELIPE MOREIRA LIMA
MARTA NÓBREGA MARTINEZ
NEWTON JOSÉ NOGUEIRA DE CASTRO
PAULO RUBENS PEREIRA DINIZ
ROBERTO CHABO
RONAN TANUS
TEXTOS REVISADOS DA 4ª EDIÇÃO E DE EDIÇÕES ANTERIORES
Monografias
Ácido ascórbico (129)
Ágar (130)
Alopurinol (132)
Amoxicilina triidratada (76)
Amoxicilina triidratada cápsulas (76.1)
Amoxicilina triidratada pó para suspensão oral (76.2)
Ampicilina (77)
Ampicilina cápsulas (77.1)
Ampicilina comprimidos (77.2)
Ampicilina pó para suspensão oral (77.3)
Ampicilina sódica (78)
Ampicilina sódica pó para solução injetável (78.1)
Ampicilina triidratada (79)
Ampicilina triidratada cápsulas (79.1)
Ampicilina triidratada comprimidos (79.2)
Ampicilina triidratada pó para suspensão injetável (79.3)
Ampicilina triidratada pó para suspensão oral (79.4)
Benzilpenicilina benzatina (82)
Benzilpenicilina benzatina estéril pó para suspensão injetável
(82.1)
Benzilpenicilina potássica (83)
Benzilpenicilina potássica estéril pó para solução injetável (83.1)
Benzilpenicilina procaína (84)
Benzilpenicilina procaína estéril pó par suspensão injetável (84.1)
Benzilpenicilina sódica (85)
Benzilpenicilina sódica estéril pó para solução injetável (85.1)
Carbonato de lítio (135)
Cloreto de potássio (138)
Cloreto de sódio (139)
Cloridrato de propranolol (143)
Dipirona (145)
Etionamida (146)
Extratos (147)
Extratos fluidos (148)
Fluoreto de sódio (151)
Furosemida (152)
Glibenclamida (153)
Glicose (28)
Lanatosídeo C (156)
Lidocaína (157)
Macela (158)
Merbromina (160)
Metilparabeno (162)
Noz-de-cola (164)
Oxamniquina (166)
Paracetamol (167)
Permanganato de potássio (168)
Propilparabeno (169)
Soros hiperimunes para uso humano (100)
Sulfato de atropina (170)
Vacina BCG (117)
Vacina de vírus vivos contra a febre amarela (124)
Texto da Parte I
Conteúdo
Índice
NOVOS TEXTOS INCLUÍDOS NO TERCEIRO FASCÍCULO
Monografias
Albendazol (131)
Albendazol, comprimidos (131.1)
Albendazol, suspensão oral (131.2)
Alopurinol, comprimidos (132.1)
Calêndula (134)
Cimetidina (136)
Cimetidina, comprimidos (136.1)
Cimetidina, solução injetável (136.2)
Ciprofloxacino (137)
Ciprofloxacino, comprimidos (137.1)
Ciprofloxacino, solução injetável (137.2)
Ciprofloxacino, solução oftálmica (137.3)
Cloridrato de biperideno (140)
Cloridrato de biperideno, comprimidos (140.1)
Cloridrato de ciprofloxacino (141)
Cloridrato de metoclopramida (142)
Cloridrato de propranolol, comprimidos (143.1)
Diclofenaco sódico (144)
Diclofenaco sódico, comprimidos (144.1)
Dipirona, comprimidos (145.1)
Dipirona, solução injetável (145.2)
Dipirona, solução oral (145.3)
Etionamida, comprimidos (146.1)
Extratos moles (149)
Extratos secos (150)
Furosemida, comprimidos (152.1)
Glibenclamida, comprimidos (153.1)
Ibuprofeno (155)
Ibuprofeno, comprimidos (155.1)
Mebendazol (159)
Mebendazol, suspensão oral (159.1)
Mesilato de pefloxacino (161)
Norfloxacino (163)
Norfloxacino, comprimidos (163.1)
Ofloxacino (165)
Ofloxacino, comprimidos (165.1)
Ofloxacino, solução injetável (165.2)
Paracetamol, comprimidos (167.1)
Paracetamol, solução oral (167.2)
Pefloxacino, comprimidos (161.1)
Sulfato de atropina, solução injetável (170.1)
Zidovudina (171)

MONOGRAFIAS
129
ÁCIDO ASCÓRBICO
Acidum ascorbicum
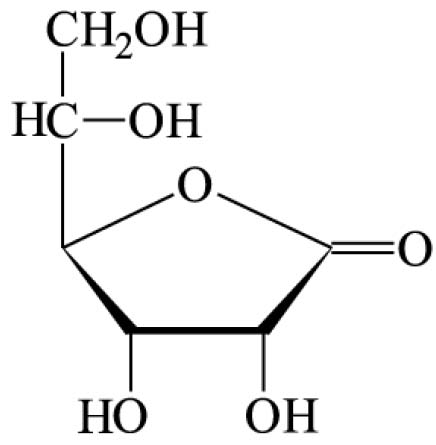
C6H8O6
176,13
0074.01-2
Ácido L-ascórbico
Contém, no mínimo, 99,0% e, no máximo, 100,5% de C6H8O6, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó fino cristalino branco ou ligeiramente amarelo, inodoro e de sabor ácido. No estado sólido é estável ao ar, mas em solução oxida-se rapidamente. Sua solução aquosa é límpida.
Solubilidade. Facilmente solúvel em água, solúvel em etanol e em acetona, insolúvel em éter etílico, clorofórmio, éter de petróleo e benzeno.
Constantes físico-químicas
Faixa de fusão (V.2.2): 189 oC a 192 ºC, com decomposição.
Poder rotatório específico (V.2.8): + 20,5º a + 21,5º, determinado em solução aquosa a 10% (p/V).
IDENTIFICAÇÃO
A. A uma alíquota de solução a 2% (p/V) adicionar solução de tartarato cúprico alcalino SR e deixar à temperatura ambiente. Observa- se mudança de coloração devido à redução lenta do tartarato cúprico. Sob aquecimento a redução é mais rápida.
B. A 2 ml de solução a 2% (p/V) adicionar 4 gotas de solução etanólica a 0,05% (p/V) de azul de metileno e aquecer a 40 ºC. A cor azul intensa torna-se mais clara ou é completamente descorada em 3 minutos.
C. Dissolver 15 mg da amostra em 15 ml de solução de ácido tricloroacético a 5% (p/V), adicionar cerca de 0,2 g de carvão ativado e agitar vigorosamente por um minuto. Filtrar até limpidez. A 5 ml do filtrado, adicionar 1 gota de pirrol, agitar levemente até completa dissolução e aquecer em banho-maria a 50 ºC. Desenvolve-se coloração azul.
ENSAIOS DE PUREZA
pH (V.2.19). 2,2 a 2,5. Determinar em solução aquosa a 5% (p/V).
Metais pesados (V.3.2.3 - Método I). Dissolver 1 g em 25 ml de água.
No máximo 0,002% (20 ppm).
Perda por dessecação (V.2.9). Determinar em 1 g de amostra, em dessecador a vácuo sobre ácido sulfúrico por 24 horas. No máximo 0,4%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,1%.
DOSEAMENTO
Pesar, exatamente, cerca de 0,2 g da amostra e dissolver em mistura de 100 ml de água isenta de dióxido de carbono e 25 ml de ácido sulfúrico 10% (p/V). Acrescentar 3 ml de amido SI e titular imediatamente com iodo 0,05 M SV. Cada ml de iodo 0,05 M SV equivale a 8,806 mg de C6H8O6.
EMBALAGEM E ARMAZENAMENTO
Em recipientes herméticos e opacos.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Vitamina.
________________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Tartarato cúprico alcalino SR (solução de Fehling)
Preparação - Misturar volumes iguais das soluções A e B, preparadas como descrito a seguir.
Solução A: dissolver 34,66 g de pequenos cristais de sulfato cúprico, cuidadosamente selecionados, sem traços de eflorescência ou umidade aderente, em água para 500 ml. Acondicionar em recipientes pequenos e herméticos.
Solução B: dissolver 173 g de tartarato de sódio e potássio cristalizado e 50 g de hidróxido de sódio, em água para 500 ml. Acondicionar em recipientes pequenos e resistentes a álcalis.
130
ÁGAR
Agar
Substância seca, coloidal, hidrofílica extraída de algas Gelidium cartilaginum L. (Gaillon) -GELIDIACEAE, Gracilaria confervoides L. (Greville) - SPHAEROCOCCACEAE e algas vermelhas relacionadas (Classe RHODOPHYCEAE).
DESCRIÇÃO MACROSCÓPICA
Apresenta-se em feixes, consistindo de tiras membranosas aglutinadas ou em formas granuladas, floculadas ou cortadas. Pode apresentar-se com a coloração amarelo-alaranjada, cinza-amarelada, levemente amarela ou incolor. É resistente quando úmido e quebradiço quando seco.
DESCRIÇÃO MICROSCÓPICA
Em montagem na água, o ágar apresenta-se granular e um tanto filamentoso; fragmentos de espícula de espongiários e algumas frústulas de diatomáceas podem estar presentes. Eventualmente, conforme a procedência, podem estar presentes frústulas de Arachnoidiscus ehrenbergii Baillon, que se caracterizam pela forma de disco de 100 a 300 μm de diâmetro.
DESCRIÇÃO MICROSCÓPICA DO PÓ
O ágar pulverizado apresenta cor branca ou branca-amarelada ou levemente amarela. Em montagem em cloral hidratado os seus fragmentos são transparentes, mais ou menos granulares, estriados e angulares, podendo, ocasionalmente, conter frústulas de diatomáceas.
IDENTIFICAÇÃO
A. Dissolver 0,1 g da amostra sob aquecimento, em 50 ml de água.
Esfriar. A 1 ml da mucilagem, adicionar, cuidadosamente, 3 ml de água, de modo a formar duas camadas distintas. Adicionar 0,1 ml de solução de iodo 0,05 M. Desenvolve-se, na interface, coloração castanho- violeta. Agitar a mistura. O líquido adquire coloração amarela pálida.
B. Adicionar 0,5 ml de ácido clorídrico a 5 ml da mucilagem obtida no teste A de Identificação. Aquecer por 30 minutos em banho-maria. Adicionar 1 ml de solução de cloreto de bário 0,67% (p/V). Desenvolve- se turvação branca após 30 minutos.
C. Aquecer 0,5 g da amostra com 50 ml de água em banho-maria, até dissolução. Apenas uns poucos fragmentos permanecem insolúveis. Ao resfriar, a solução geleifica entre 35 oC e 30 oC. Aquecer o gel obtido em banho-maria. Não ocorre liquefação em temperatura inferior a 85 oC.
ENSAIOS DE PUREZA
Índice de intumescência (V.4.2.13). Determinar sobre amostra pulverizada (355 μm). O índice de intumescência não é inferior a 10 e difere de, no máximo, 10% do valor declarado no rótulo.
Matérias estranhas insolúveis. Pesar 5 g da amostra pulverizada (tamiz
44), adicionar 100 ml de água e 14 ml de ácido clorídrico
diluído. Ferver, cuidadosamente, por 15 minutos, agitando frequentemente.
Filtrar o líquido quente através de um funil de vidro sinterizado, previamente tarado, lavar o filtro com água quente e secar entre 100 oC e 105 oC. No máximo 1%.
Gelatina. Pesar 1 g da amostra, adicionar 100 ml de água e aquecer em banho-maria até dissolução. Deixar esfriar até 50 oC. A 5 ml da solução anterior, adicionar 5 ml de ácido pícrico a 10% (p/V). Não aparece turbidez após 10 minutos.
Determinação de água (V.4.2.3). Determinar em 1 g da amostra pulverizada (tamiz 44), em estufa, entre 100 oC e 105 oC, até peso constante. No máximo 20%.
Cinzas totais (V.4.2.4). No máximo 5%, em relação à substância dessecada.
TESTES DE SEGURANÇA BIOLÓGICA
Contagem de microrganismos viáveis totais (V.5.1.6.1). Determinar pelo método de contagem em placa. No máximo 1 000 UFC/g.
Pesquisa e identificação de patógenos (V.5.1.7). Ausência de Escherichia coli e Salmonella sp.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, ao abrigo da umidade.
ROTULAGEM
Observar a legislação vigente.
CATEGORIA
Adjuvante (espessante).
131
ALBENDAZOL
Albendazolum
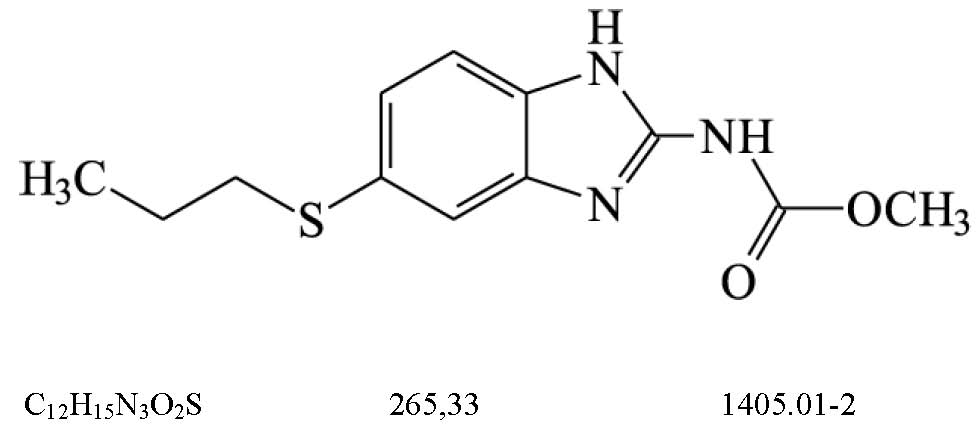
(5-propiltio-1H-benzimidazol-2-il)carbamato de metila
Contém, no mínimo, 98,0% e, no máximo, 102,0% de C12H15N3O2S, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó cristalino, untuoso ao tato, branco, quase inodoro.
Solubilidade. Praticamente insolúvel em água, facilmente solúvel emácido fórmico, solúvel em ácido acético glacial e ácido sulfúrico, pouco solúvel em clorofórmio, muito pouco solúvel em acetato de etila, acetona, álcool terc-butílico, benzeno, cloreto de metileno, etanol,éter etílico, isopropanol, metanol, tolueno, insolúvel em n-hexano e tetracloreto de carbono. Muito pouco solúvel em ácido clorídrico 0,1 M e insolúvel em hidróxido de sódio 0,1 M.
Constantes físico-químicas
Faixa de fusão (V.2.2): 208 °C a 209 °C.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) de amostra dessecada até peso constante, e dispersa em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de albendazol padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de clorofórmio-ácido acético glacial-éter (60:10:10), como fase móvel.
Aplicar, separadamente, à placa 10 μl de cada uma das soluções recentemente preparadas, descritas a seguir.
Solução (1): solução a 1% (p/V) de amostra em ácido acético glacial.
Solução (2): solução a 1% (p/V) de albendazol padrão em ácido acético glacial.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e examinar sob luz ultravioleta (254 nm). A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidade, àquela obtida com a solução (2).
C. Dissolver, em tubo de ensaio, 10 mg de amostra em 5 ml de
clorofórmio. Transferir 1 ml para tubo de ensaio contendo 5 ml de
ácido sulfúrico e 4 gotas de formaldeído. Desenvolve-se coloração na
interface. Após agitação a camada sulfúrica rica também desenvolve
coloração.
ENSAIOS DE PUREZA
Perda por dessecação (V.2.9). Determinar em 2 g de amostra, em estufa a 105 ºC, por 4 horas. No máximo 0,5%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,2%.
DOSEAMENTO
A. Por titulação em meio não-aquoso (V.3.4.5). Utilizar 0,4 g de amostra, dessecada em estufa a 105 °C, por 3 horas e dissolver em 30 ml de ácido acético glacial. Aquecer, se necessário. Esfriar e adicionar 5 gotas de cloreto de metilrosanilínio (cristal violeta) a 0,1% (p/V) em ácido acético glacial. Titular com ácido perclórico 0,1 M SV, até coloração verde esmeralda. Realizar ensaio em branco e fazer as correçõesnecessárias. Cada ml de ácido perclórico 0,1 M SV equivale a 26,53 mg de C12H15N3O2S.
B. Por espectrofotometria de absorção no ultravioleta (V.2.14-3). Pesar, exatamente, cerca de 25 mg de amostra e dissolver em 25 ml deácido clorídrico a 2% (V/V) em metanol. Completar o volume para 50 ml com água. Transferir 5 ml para balão volumétrico de 50 ml e completar o volume com ácido clorídrico 0,1 M. Diluir, sucessivamente, em hidróxido de sódio 0,1 M, até a concentração final de 0,0005% (p/V). Preparar solução padrão na mesma concentração, utilizando os mesmos solventes. Medir as absorvâncias das soluções resultantes em 309 nm, utilizando hidróxido de sódio 0,1 M como branco. Calcular o teor de C12H15N3O2S na amostra, a partir das leituras obtidas.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Anti-helmíntico.
________________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Ácido pícrico
Sinonímia - 2,4,6-Trinitrofenol.
Fórmula e massa molecular: C6H3N3O7 - 229,1.
Descrição - Cristais amarelos, solúveis em água e em álcool.
Conservação - Armazenar umedecido com água.
131.1
ALBENDAZOL COMPRIMIDOS
Contém, no mínimo 90,0% e, no máximo, 110,0% da quantidade declarada de C12H15N3O2S.
IDENTIFICAÇÃO
A. Pesar e pulverizar os comprimidos. Transferir quantidade de pó equivalente a 10 mg de albendazol para balão volumétrico de 50 ml e adicionar 25 ml de ácido clorídrico a 2% (V/V) em metanol. Agitar por 10 minutos, completar o volume com o mesmo solvente e filtrar. Diluir o filtrado até concentração de 0,001% (p/V) com o mesmo solvente. Preparar solução padrão na mesma concentração. O espectro de absorção no ultravioleta (V.2.14.-3), da solução amostra, na faixa de 200 a 400 nm, exibe máximos e mínimos somente nos mesmos comprimentos de onda da solução padrão.
B. Proceder conforme descrito no método B de Doseamento. O tempo de retenção do pico principal obtido no cromatograma com a solução amostra corresponde àquele do pico principal do cromatograma obtido com a solução padrão.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). No máximo 15 minutos.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: ácido clorídrico 0,1 M, 900 ml
Aparelhagem: pás, 50 rpm
Tempo: 30 minutos
Procedimento: após o teste, filtrar e retirar alíquota de 10 ml do meio de dissolução e transferir para balão volumétrico de 250 ml, completando o volume com hidróxido de sódio 0,1 M. Transferir 90 mg de padrão de albendazol para balão volumétrico de 250 ml, adicionar 10 ml de ácido clorídrico a 2% (V/V) em metanol e homogeneizar.Diluir com ácido clorídrico 0,1 M até completar o volume. Transferir 5 ml desta solução para balão volumétrico de 200 ml e diluir com hidróxido de sódio 0,1 M. Medir as absorvâncias em 308 e 350 nm, utilizando o mesmo solvente para o ajuste do zero. Calcular a quantidade de C12H15N3O2S dissolvido no meio pela expressão: 22,5 C (Aa/Ap), em que C é a concentração, em μg/ml, de albendazol na solução padrão e Aa e Ap são as diferenças entre as absorvâncias a 308 e 350 nm, obtidas para a solução amostra e para a solução padrão, respectivamente.Tolerância: não menos do que 80% (T) da quantidade declarada de C12H15N3O2S se dissolvem em 30 minutos.
DOSEAMENTO
A. Por espectrofotometria de absorção no ultravioleta (V.2.14.-3).
Pesar e pulverizar 20 comprimidos. Transferir quantidade de pó equivalente
a 10 mg de albendazol para balão volumétrico de 50 ml e
adicionar 25 ml de ácido clorídrico a 2% (V/V) em metanol. Agitar
por 10 minutos, completar o volume com água destilada e filtrar.
Diluir, sucessivamente, até a concentração de 0,0008% (p/V), utilizando
hidróxido de sódio 0,1 M como solvente. Preparar solução
padrão na mesma concentração, utilizando os mesmos solventes. Medir
as absorvâncias das soluções em 308 nm, utilizando hidróxido de
sódio 0,1 M para o ajuste do zero. Calcular o teor de C12H15N3O2S na
amostra, a partir das leituras obtidas.
B. Por cromatografia líquida de alta eficiência (V.2.17.4). Utilizar cromatógrafo provido de detector a 254 nm, coluna de 250 mm de comprimento e 4,6 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octadecilsilano (5 μm); fluxo da fase móvel de 2 ml/minuto.
Fase móvel: solução de 0,5 g de fosfato de amônio monobásico em 400 ml de água-600 ml de metanol.
Solução amostra: pesar e pulverizar 20 comprimidos. Transferir quantidade
de pó equivalente a 100 mg de albendazol para balão volumétrico
de 50 ml, adicionar 5 ml de ácido sulfúrico 1% (V/V) em
metanol e 20 ml de metanol. Agitar por 15 minutos, completar o
volume com metanol e filtrar. Transferir 5 ml do filtrado e 5 ml da
solução de padrão interno para balão volumétrico de 50 ml e completar
o volume com metanol.
Solução padrão: pesar, exatamente, cerca de 100 mg de albendazol padrão e transferir para balão volumétrico de 50 ml, adicionar 5 ml deácido sulfúrico 1% (V/V) em metanol e completar o volume com metanol. Transferir 5 ml desta solução e 5 ml da solução de padrão interno para balão volumétrico de 50 ml e completar o volume com o metanol. Solução de padrão interno: pesar, exatamente, cerca de 150 mg de parbendazol. Transferir para balão volumétrico de 50 ml, adicionar 5 ml de ácido sulfúrico 1% (V/V) em metanol e completar o volume com metanol.
A eficiência da coluna não deve ser menor que 4 000 pratos teóricos/metro. A resolução entre albendazol e parbendazol não deve ser menor que 2. O desvio padrão relativo das áreas de replicatas dos picos registrados não deve ser maior que 2,0%. Procedimento: injetar separadamente 20 μl das soluções amostra e padrão, registrar os cromatogramas e medir as áreas dos picos. Calcular a quantidade de C12H15N3O2S na solução amostra a partir das respostas obtidas para solução padrão e amostra em relação a solução de padrão interno.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
131.2
ALBENDAZOL SUSPENSÃO ORAL
Albendazol suspensão oral é mistura de albendazol com um ou mais agentes corantes, aromatizantes, tampões, adoçantes e conservantes, em veículo aquoso. Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C12H15N3O2S.
IDENTIFICAÇÃO
Diluir volume adequado da suspensão em mistura de metanol e ácido clorídrico (99:1) para obter concentração de 1 mg/ml. Filtrar, se necessário, transferir 1 ml do filtrado para balão volumétrico de 100 ml, completar o volume com hidróxido de sódio 0,1 M e homogeneizar. O espectro de absorção da solução no ultravioleta (V.2.14.- 3), na faixa de 200 a 400 nm, exibe máximos e mínimos somente nos mesmos comprimentos de onda de solução similar de albendazol padrão.
CARACTERÍSTICAS
Determinação de volume (V.1.2). Cumpre o teste.
pH (V.2.19). 4,5 a 5,5.
DOSEAMENTO
Proceder onforme descrito em Cromatografia líquida de alta eficiência (V.2.17.4), utilizando cromatógrafo provido de detector a 308 nm, coluna de 250 mm de comprimento e 4 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octadecilsilano; fluxo da fase móvel de 2 ml/minuto. Fase móvel: dissolver 11 g de fosfato de sódio monobásico em 800 ml de água e adicionar 1 200 ml de metanol. Solução amostra: transferir para balão volumétrico de 100 ml volume da suspensão correspondente a 100 mg de albendazol e completar o volume com mistura de metanol e ácido clorídrico (99:1). Diluir, sucessivamente, até a concentração de 100 μg/ml, utilizando fase móvel como solvente. Filtrar, se necessário. Solução padrão: pesar, exatamente, cerca de 50 mg de albendazol padrão. Transferir para balão volumétrico de 50 ml e completar o volume com a mistura de metanol e ácido clorídrico (99:1). Diluir, sucessivamente, até a concentração de 100 μg/ml, utilizando fase móvel como solvente. A eficiência da coluna não deve ser menor que 8 000 pratos teóricos/metro. O desvio padrão relativo das áreas de replicatas dos picos registrados não deve ser maior que 2%. Procedimento: injetar, separadamente, 20 μl das soluções amostra e padrão, registrar os cromatogramas e medir as áreas dos picos. Calcular a quantidade de C12H15N3O2S na solução amostra a partir das respostas obtidas para solução padrão e amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, a temperatura ambiente.
ROTULAGEM
Observar a legislação vigente.
132
ALOPURINOL
Allopurinolum
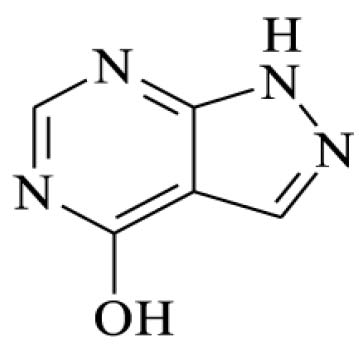
C5H4N4O 1 3 6 , 11 0031.01-1
1,5-Diidro-4H-pirazolo[3,4-d]pirimidin-4-ona
Contém, no mínimo, 98,0% e, no máximo, 101,0% de C5H4N4O, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó leve, branco ou quase branco e praticamente inodoro.
Solubilidade. Muito pouco solúvel em água e em álcool, praticamente insolúvel em clorofórmio e em éter etílico. Solúvel em soluções aquosas de hidróxido de sódio e potássio.
IDENTIFICAÇÃO
A. O espetro de absorção no infravermelho (V.2.14.-4) da amostra dessecada até peso constante, e dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de alopurinol padrão, preparado de maneira idêntica.
B. Dissolver cerca de 50 mg da amostra em 10 ml de hidróxido de sódio 0,1 M, aquecer se necessário, resfriar e adicionar água para completar 50 ml. Diluir 1 ml com ácido clorídrico 0,1 M para 100 ml. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 200 nm a 350 nm, da solução final exibe máximos e mínimos somente nos mesmos comprimentos de onda de solução similar de alopurinol padrão. A razão entre os valores de absorvância medidos em 231 nm e 250 nm está compreendida entre 0,50 e 0,62.
C. Proceder conforme descrito em Substâncias relacionadas. A mancha principal obtida no cromatograma com a solução (1) corresponde em tamanho, cor e posição à mancha obtida no cromatograma com a solução (3).
D. Dissolver 0,3 g da amostra em 2,5 ml de hidróxido de sódio 2 M, adicionar 50 ml de água e, lentamente, sob agitação, 5 ml de solução de nitrato de prata a 4,25% (p/V). Forma-se um precipitado branco, que não se dissolve após adição de 5 ml de amônia 10 M.
E. Dissolver 50 mg da amostra em 5 ml de hidróxido de sódio 2 M, adicionar 1 ml de solução alcalina de tetraiodomercurato de potássio, aquecer até ebulição e deixar em repouso. Forma-se um precipitado amarelo e floculento.
ENSAIOS DE PUREZA
Limpidez da solução (IV.-3). A solução a 5% (p/V) em hidróxido de sódio 2 M é límpida.
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando celulose com indicador de fluorescência, como suporte. Agitar 200 ml de álcool nbutílico com 200 ml de hidróxido de amônio 6 M e desprezar a fase inferior. Utilizar como fase móvel a fase superior, adicionada de 20 ml de álcool n-butílico. Aplicar, separadamente, à placa, 10 μl de cada uma das soluções descritas a seguir, recentemente preparadas.
Solução (1): solução a 2,5% (p/V) da amostra em mistura de hidróxido de amônio 6 M e hidróxido de sódio M (9:1).
Solução (2): solução a 0,005% (p/V) de hemissulfato de 3-amino-4- carboxamidopirazol em mistura de hidróxido de amônio 6 M e hidróxido de sódio M (9:1).
Solução (3): solução a 2,5% (p/V) de alopurinol padrão em mistura de hidróxido de amônio 6 M e hidróxido de sódio M (9:1).
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e examinar sob luz ultravioleta (254 nm). Nenhuma mancha secundária obtida no cromatograma com a solução (1) é mais intensa que a mancha obtida com a solução (2).
Perda por dessecação (V.2.9). Determinar em 1 g de amostra, em estufa a 105 oC, até peso constante. No máximo 0,5%. Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,1%.
DOSEAMENTO
A. Por titulação em meio não-aquoso. Pesar, exatamente, cerca de 0,2 g de amostra e dissolver em 50 ml de dimetilformamida previamente tratada. Adicionar três gotas de solução metanólica a 0,3% (p/V) de azul de timol SI, recentemente preparada. Titular com metóxido de sódio 0,1 M SV até viragem para azul. Cada ml de metóxido de sódio 0,1 M SV equivale a 13,61 mg de C5H4N4O.
B. Por espectrofotometria de absorção no ultravioleta. Dissolver, exatamente, cerca de 50 mg da amostra em 10 ml de solução de hidróxido de sódio 0,1 M e aquecer, se necessário. Resfriar, completar o volume para 50 ml com água e homogeneizar. Diluir quantitativamente com ácido clorídrico 0,1 M para obter concentração de 0,001% (p/V). Preparar solução padrão na mesma concentração, utilizando os mesmos solventes da solução amostra. Medir as absorvâncias das soluções resultantes em 250 nm (V.2.14.-3), utilizandoácido clorídrico 0,1 M para ajuste do zero. Calcular o teor de C5H4N4O na amostra, a partir das leituras obtidas. Alternativamente, realizar o cálculo utilizando A(1%,1cm) = 563, em 250 nm.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antigotoso.
________________________________________________________
XII.2 REAGENTES E SOLUÇÕES REAGENTES
Solução alcalina de tetraiodomercurato de potássio
Preparação - Dissolver em água 11 g de iodeto de potássio e 15 g de iodeto de mercúrio (II) e diluir com água para 100 ml. Imediatamente antes do uso, misturar a soluçãoanterior cm igual volume de hidróxido de sódio a 25% (p/V).
132.1
ALOPURINOL COMPRIMIDOS
Contém, no mínimo, 92,5% e, no máximo, 107,5% da quantidade declarada de C5H4N4O.
IDENTIFICAÇÃO
A. Pesar e pulverizar os comprimidos. Misturar em gral quantidade de pó equivalente a 50 mg de alopurinol com 10 ml de hidróxido de sódio 0,1 M. Filtrar, acidificar o filtrado com ácido acético 1 M, coletar o precipitado, lavar com porções de 3 ml de etanol absoluto e, finalmente, com 4 ml de éter etílico anidro. Deixar secar ao ar por 15 minutos e posteriormente dessecar a 105 oC por 3 horas. O espectro de absorção no infravermelho (V.2.14.-4) do resíduo, disperso em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro do alopurinol padrão, disperso em brometo de potássio.
B. A solução amostra obtida no Doseamento responde ao teste B de Identificação descrito na monografia de alopurinol.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: ácido clorídrico 0,1 M, 900 ml
Aparelhagem: pás, 75 rpm
Tempo: 45 minutos
Procedimento: após o teste, retirar volume suficiente do meio de dissolução, filtrar e diluir em ácido clorídrico 0,1 M até concentração adequada. Medir as absorvâncias das soluções em 250 nm (V.2.14.-3), utilizando o mesmo solvente para ajuste do zero. Calcular a quantidade de C5H4N4O dissolvido no meio, comparando as leituras obtidas com a da solução padrão na concentração de 0,001% (p/V), preparada no mesmo solvente.
Tolerância: não menos que 75% (T) da quantidade declarada de C5H4N4O se dissolvem em 45 minutos.
DOSEAMENTO
Pesar e pulverizar 20 comprimidos. Transferir, para balão volumétrico de 100 ml, quantidade exatamente pesada do pó equivalente a 0,1 g de alopurinol. Adicionar 20 ml de hidróxido de sódio 0,1 M, submeter ao ultra-som ou agitar mecanicamente por 15 minutos, completar o volume com água e homogeneizar. Filtrar em papel de filtro adequado, desprezando os primeiros mililitros do filtrado e diluir comácido clorídrico 0,1 M até concentração de 0,001% (p/V). Pesar, exatamente, cerca de 50 mg de alopurinol padrão, dissolver em 10 ml de hidróxido de sódio 0,1 M e diluir com água para 50 ml. Homogeneizar e diluir quantitativamente com ácido clorídrico 0,1 M para obter concentração de 0,001% (p/V).Medir as absorvâncias das soluções resultantes em 250 nm (V.2.14.-3), utilizando ácido clorídrico 0,1 M para ajuste do zero. Calcular o teor de C5H4N4O nos comprimidos a partir das leituras obtidas. Alternativamente, realizar o cálculo utilizando A(1%, 1cm)=563, em 250 nm.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
133
BICARBONATO DE SÓDIO
Natrii hidrogenocarbonas
NaHCO3 84,00 1113.03- 8
Contém, no mínimo, 99,0% e, no máximo, 101,0% de NaHCO3.
DESCRIÇÃO
Caracteres físicos. Pó branco, cristalino, inodoro. Quando aquecido, seco ou em solução, converte-se, gradativamente, em carbonato de sódio.
Solubilidade. Solúvel em água, praticamente insolúvel em etanol.
IDENTIFICAÇÃO
A. Preparar solução de bicarbonato de sódio a 5% (p/V) em água isenta de dióxido de carbono. A 5 ml desta solução adicionar 0,1 ml de solução de fenolftaleína SI. Desenvolve-se coloração rósea. Aquecer.
Ocorre liberação de gás e a coloração da solução muda para vermelho.
B. Responde às reações de íons carbonato e bicarbonato (V.3.1.1).
C. Responde às reações do íon sódio (V.3.1.1).
ENSAIOS DE PUREZA
Aspecto da solução. A solução descrita no teste A de Identificação é límpida e incolor.
Carbonatos. O pH da solução descrita no teste A de Identificação, recém-preparada, não é superior a 8,6.
Cloreto (V.3.2.1). A 7 ml da solução descrita no teste A de Identificação, adicionar 2 ml de ácido nítrico e diluir para 15 ml comágua. Prosseguir conforme descrito em Ensaio-limite para cloretos. No máximo 0,015% (150 ppm). Sulfato (V.3.2.2). Suspender 1 g da amostra em 10 ml de água e adicionar ácido clorídrico até a neutralidade. Prosseguir conforme descrito em Ensaio-limite para sulfatos. No máximo 0,015% (150 ppm).Amônia (V.3.2.6). Diluir 10 ml da solução descrita no teste A de Identificação para 15 ml com água. Prosseguir conforme descrito em Ensaio-limite para amônia. No máximo 0,002% (20 ppm). Arsênio (V.3.2.5). A 0,5 g de amostra, adicionar ácido sulfúrico 3,5 M até cessar a efervescência. Prosseguir conforme descrito em Ensaio-limite para arsênio. No máximo 0,0002% (2 ppm). Cálcio (V.3.2.7). Neutralizar a suspensão de 1 g da amostra em 10 ml de água com ácido clorídrico e diluir para 15 ml com água. Prosseguir conforme descrito em Ensaio-limite para cálcio. No máximo 0,01% (100 ppm). Metais pesados (V.3.2.3 - Método II). Dissolver 2 g da amostra na mistura de 2 ml de ácido clorídrico e 18 ml de água. Utilizar 12 ml da solução e prosseguir conforme descrito em Ensaio-limite para metais pesados. No máximo 0,001% (10 ppm). Ferro (V.3.2.4). Dissolver 0,5 g em 5 ml de ácido clorídrico. Prosseguir conforme descrito em Ensaio-limite para ferro. No máximo 0,002% (20 ppm).
DOSEAMENTO
Dissolver 1,5 g da amostra em 50 ml de água isenta de dióxido de
carbono. Titular com ácido clorídrico M SV, utilizando 0,2 ml de
alaranjado de metila SI como indicador. Cada ml de ácido clorídrico
M SV corresponde a 84 mg de NaHCO3.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antiácido.
134
CALÊNDULA
Calendulae flos
Calendula officinalis L. - ASTERACEAE
A droga vegetal consiste de flores liguladas inteiras ou trituradas, acompanhadas de escassas flores tubulosas, separadas do receptáculo e das brácteas involucrais, secas. Não deve conter menos que 0,4% de flavonóides totais, calculados como hiperosídeo (C21H20O12) em relação ao material dessecado.
NOMES POPULARES
Maravilha.
CARACTERES ORGANOLÉPTICOS
A droga possui odor fraco e sabor levemente amargo.
DESCRIÇÃO MACROSCÓPICA
Flores dispostas em capítulos de 3 cm a 7 cm, envolvidas por um invólucro de 2 séries de brácteas. As flores da periferia são liguladas, pistiladas, de 1,5 cm a 3,0 cm de comprimento e 0,5 cm a 0,7 cm de largura na porção mediana da lígula. Corolas amareladas ou alaranjadas, com o limbo tridentado, apresentando 4 ou 5 nervuras e tubo curto coberto de tricomas, ocasionalmente acompanhadas de um estilete filiforme e um estigma bífido. As flores do centro são escassas, tubulosas, pequenas, curtas, de aproximadamente 0,5 cm de comprimento, hermafroditas, amarelas ou alaranjadas, raro quase avermelhadas, com corola quinqüedentada; anteras sagitadas e estilete indiviso. Papus ausente.
DESCRIÇÃO MICROSCÓPICA
Em vista frontal, a face adaxial da epiderme da corola ligulada mostra células retangulares, alongadas, de contorno levemente sinuoso, com cutícula estriada e é destituída de estômatos. Na região apical desta mesma face, as células são menores e arranjadas menos regularmente; no extremo basal da lígula existe uma camada de células com espessamento nas paredes externas contendo prismas e pequenos aglomerados de cristais. A face abaxial da epiderme é semelhante à adaxial, diferindo desta por apresentar poucos estômatos anomocíticos, os quais são relativamente grandes na região apical da lígula, quando comparados com as demais células epidérmicas desta porção.
Na região basal da face abaxial ocorrem tricomas tectores longos, multicelulares, bisseriados, cônicos, de ápice arredondado e tricomas glandulares multicelulares, de pedicelo unisseriado, com 3 a 5 células, ou bisseriado, com 3 ou 4 células em cada fileira, ambos com cabeça ovalada, multicelular, geralmente bisseriada. As células do parênquima subjacente da corola ligulada apresentam numerosas gotas deóleo de coloração amarelo-alaranjada a amarelo-clara. O parênquima da lígula é atravessado longitudinalmente por 4 ou 5 feixes vasculares, com elementos de vaso apresentando espessamentos anelados e helicoidais. Junto às células parenquimáticas das corolas tubulosas são encontrados 5 feixes vasculares bifurcados abaixo da zona de soldadura das pétalas. Nas brácteas involucrais, quando presentes, ocorrem tricomas tectores longos, multicelulares, bisseriads, cônicos, de ápice arredondado, e tricomas tectores com 4 ou 5 células, unisseriadas, das quais a célula apical é muito mais longa do que as demais e freqüentemente dobrada e achatada, além de tricomas glandulares mais raros, multicelulares, de pedicelo bisseriado, cônico, com células basais mais longas e irregulares do que as demais. Nas anteras observa-se o endotécio, composto de células ligeiramente alongadas que, em vista frontal, mostram espessamentos característicos, restritos às paredes transversais (anticlinais). Associados ao endotécio, ocorrem esclereídes pequenos, alaranjados, com paredes pouco espessadas e numerosas pontoações. Os grãos de pólen são equinados, tricolpados, medindo em torno de 45 μm de diâmetro. As células epidérmicas dos estigmas são poligonais a levemente alongadas em vista frontal e mostram papilas curtas, bulbosas, enquanto as dos ovários são pequenas, poligonais em vista frontal, contendo pigmentos castanhos. Nos ovários ocorrem tricomas glandulares iguais aos das corolas liguladas. Os aquênios, quando presentes, têm forma navicular, com ornamentações dentadas na face dorsal.
DESCRIÇÃO MICROSCÓPICA DO PÓ
O pó deve atender a todas as exigências estabelecidas para a espécie, menos os caracteres macroscópicos. São característicos: a cor castanho- amarelada; presença de tubos das flores liguladas; partes de lígulas; fragmentos da epiderme das lígulas com cutícula estriada; fragmentos de parênquima subepidérmico com gotas de óleo; células basais das corolas contendo cristais; fragmentos de tecido vascular; corolas das flores tubulosas; anteras das flores tubulosas; fragmentos de anteras na maioria das vezes com porções de feixes condutores; grãos de pólen equinados, tricolpados; fragmentos de células epidérmicas dos estigmas com papilas bulbosas; fragmentos de paredes de ovários com células pigmentadas; aquênios e tricomas iguais aosdescritos acima.
IDENTIFICAÇÃO
Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando cromatoplaca de sílica-gel GF254, como fase estacionária, e mistura de ácido fórmico anidro-ácido acético glacialágua- acetato de etila (11:11:27:100), como fase móvel. Aplicar na cromatoplaca, separadamente, em forma de banda, 30 μl da solução amostra e 10 μl da solução de referência, preparados como segue.
Solução amostra: ferver sob refluxo 1 g da droga pulverizada com 20ml de etanol a 55% (V/V) por 20 minutos e filtrar.
Solução referência: dissolver 10 mg de rutina e 5 mg de ácido clorogênico em metanol e completar o volume com o mesmo solvente a 10 ml.
Desenvolver o cromatograma em percurso de aproximadamente 10 cm. Deixar a placa secar em capela e examiná-la sob luz ultravioleta (365 nm). O cromatograma deverá apresentar mancha de coloração marrom, na mesma altura que a obtida com a solução referência da rutina (Rf aproximadamente 0,35) e mancha azul correspondente aoácido clorogênico (Rf aproximadamente 0,55). O cromatograma obtido com a solução amostra deverá apresentar também mancha marrom (Rf aproximadamente 0,25), mancha azulada (Rf aproximadamente 0,30), duas manchas azuis correspondentes ao ácido clorogênico (Rf aproximadamente 0,55) e uma mancha vermelha junto ao fronte do solvente (Rf aproximadamente 0,95). Em seguida, nebulizar a placa com solução de difenilborato de aminoetanol a 1% (p/V) em metanol e examinar sob luz ultravioleta (365 nm). O cromatograma obtido com a solução amostra deverá apresentar mancha verde (Rf aproximadamente 0,30), mancha laranja correspondente à rutina (Rf aproximadamente 0,35), mancha verde correspondente ao ácido clorogênico (Rf aproximadamente 0,55), mancha laranja-claro (Rf aproximadamente 0,60) e mancha verde (Rf aproximadamente 0,90). O cromatograma obtido com a solução amostra pode apresentar ainda, uma mancha de fluorescência amarelada logo abaixo da mancha correspondenteà rutina e banda de fluorescência amarelada situada entre as manchas correspondentes à rutina e ao ácido clorogênico.
ENSAIOS DE PUREZA
Matéria estranha (V.4.2.2). No máximo 3%.
Determinação de água (V.4.2.3.). No máximo 12%.
Cinzas totais (V.4.2.4.). No máximo 10%.
DOSEAMENTO
Determinar o valor de Flavonóides Totais. Pesar exatamente cerca de
0,4 g da droga pulverizada (800 μm) e colocar em balão de fundo
redondo de 100 ml. Acrescentar 1ml de uma solução aquosa de
hexametilenotetramina a 0,5% (p/V), 20 ml de acetona e 2 ml de
ácido clorídrico. Aquecer em banho-maria, sob refluxo, por 30 minutos.
Filtrar a mistura em algodão, para balão volumétrico de 100
ml, retornando o resíduo da droga e o algodão ao mesmo balão de
fundo redondo, adicionado de 20 ml de acetona. Colocar em refluxo,
por 10 minutos. Após resfriamento à temperatura ambiente, filtrar a
solução para o balão volumétrico de 100 ml. Repetir a operação.
Após, completar o volume do balão volumétrico com acetona. Em funil de separação, tratar 20 ml desta solução com 20 ml de água e após, extrair com 15 ml de acetato de etila, repetindo-se por três vezes, com porções de 10 ml de acetato de etila. Reunir as fases de acetato de etila e lavá-las em funil de separação, com duas porções de 50 ml de água, transferindo a seguir para balão volumétrico de 50 ml, completando-se o volume com acetato de etila (solução-mãe SM).
Pipetar 10 ml desta solução, adicionar 1 ml do reagente de cloreto de alumínio, diluindo-se em balão volumétrico de 25 ml com solução metanólica de ácido acético a 5% (V/V). Preparar o branco diluindo 10 ml da SM para 25 ml em balão volumétrico com solução metanólica de ácido acético a 5% (V/V). Após 30 minutos, medir a absorvância da solução a 425 nm, em cubeta de 1 cm, utilizando o branco para ajuste do zero. Calcular a porcentagem de flavonóides totais segundo a fórmula:
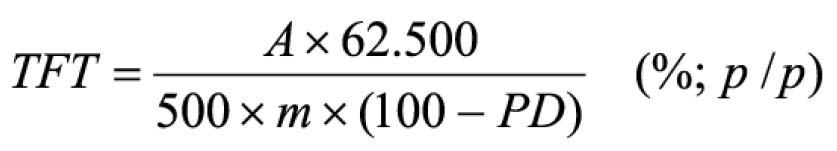
Em que
A = absorvância medida;
m = massa da droga (g);
PD= perda por dessecação (%; p/p)
O resultado é fornecido em porcentagem (p/p) de flavonóides calculados como hiperosídeo (C21H20O12).
EMBALAGEM E ARMAZENAMENTO
Em recipientes de vidro ou metal, bem-fechados, ao abrigo da luz e do calor.
________________________________________________________
XII.2 REAGENTES E SOLUÇÕES REAGENTES
Reagente de cloreto de alumínio
Dissolver 1g de cloreto de alumínio com solução metanólica de ácido acético 5% (V/V), em balão volumétrico de 50 ml. Completar o volume.
135
CARBONATO DE LÍTIO
Lithium carbonas
Li2CO3 73,89 0749.01-X
Contém, no mínimo, 98,5 % e, no máximo, 100,5 % de Li2CO3.
DESCRIÇÃO
Caracteres físicos. Pó branco.
Solubilidade. Levemente solúvel em água, praticamente insolúvel em etanol.
IDENTIFICAÇÃO
A. Quando umedecido com ácido clorídrico confere coloração vermelhaà chama não luminosa.
B. Dissolver 0,2 g em 1 ml de ácido clorídrico. Evaporar até secura em banho-maria. O resíduo dissolve em 3 ml de etanol.
C. Responde às reações do íon carbonato (V.3.1.1).
ENSAIOS DE PUREZA
Aspecto da solução. Suspender 10 g da amostra em 30 ml de água e dissolver pela adição de 22 ml de ácido nítrico. Neutralizar com solução de hidróxido de sódio SR e diluir com água para 100 ml. A solução amostra é límpida e incolor.
Cloreto (V.3.2.1). Proceder conforme descrito em Ensaio-limite para cloretos, utilizando 2,5 ml da solução obtida em Aspecto da solução.
No máximo 0,02% (200 ppm).
Sulfato (V.3.2.2). Dispersar 1,25 g da amostra em 5 ml de água e dissolver pela adição de ácido clorídrico 70% (p/V). Ferver por 2 minutos. Esfriar e adicionar solução de hidróxido de sódio SR até neutralização. Diluir para 25 ml com água. Prosseguir conforme descrito em Ensaio-limite para sulfatos. No máximo 0,02% (200 ppm).
Arsênio (V.3.2.5). A 0,5 g de amostra, adicionar ácido sulfúrico 3,5 M até cessar a efervescência e prosseguir conforme descrito em Ensaio-limite para arsênio. No máximo 0,0002% (2 ppm).
Cálcio (V.3.2.7). Utilizar 5 ml da solução obtida em Aspecto da solução. Prosseguir conforme descrito em Ensaio-limite para cálcio.
No máximo 0,002% (20 ppm).
Metais pesados (V.3.2.3 - Método II). Utilizar 10 ml da solução
obtida em Aspecto da solução e prosseguir conforme descrito em
Ensaio-limite para metais pesados. No máximo 0,002% (20 ppm).
Ferro (V.3.2.4). Utilizar 5 ml da solução obtida em Aspecto da solução e prosseguir conforme descrito em Ensaio-limite para ferro. No máximo 0,002% (20 ppm).
Magnésio (V.3.2.8). Diluir 1 ml da solução obtida em Aspecto da solução para 10 ml com água. Utilizar 6,7 ml desta solução e prosseguir conforme descrito em Ensaio-limite para magnésio. No máximo 0,015% (150 ppm).
Potássio. Dissolver 1 g da amostra em 10 ml de ácido clorídrico 70% (p/V) e diluir para 50 ml com água. Utilizar como referência, solução de cloreto de potássio contendo 0,5 mg de potássio por ml. Proceder conforme descrito em Espectrofotometria de emissão atômica (V.2.23). Medir a intensidade de emissão em 766,5 nm. No máximo 0,03% (300 ppm).
Sódio. Dissolver 1 g da amostra em 10 ml de ácido clorídrico 70% (p/V) e diluir para 50 ml com água destilada. Utilizar como referência, solução de cloreto de sódio contendo 0,5 mg de sódio por ml. Proceder conforme descrito em Espectrofotometria de emissão atômica (V.2.23). Medir a intensidade de emissão em 589 nm. No máximo 0,03% (300 ppm).
DOSEAMENTO
Dissolver 0,5 g da amostra em 25 ml de ácido clorídrico M SV.
Titular com solução de hidróxido de sódio M SV utilizando alaranjado
de metila SI como indicador. Cada ml de ácido clorídrico M
SV equivale a 36,95 mg de Li2CO3.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antidepressivo.
136
CIMETIDINA
Cimetidinum
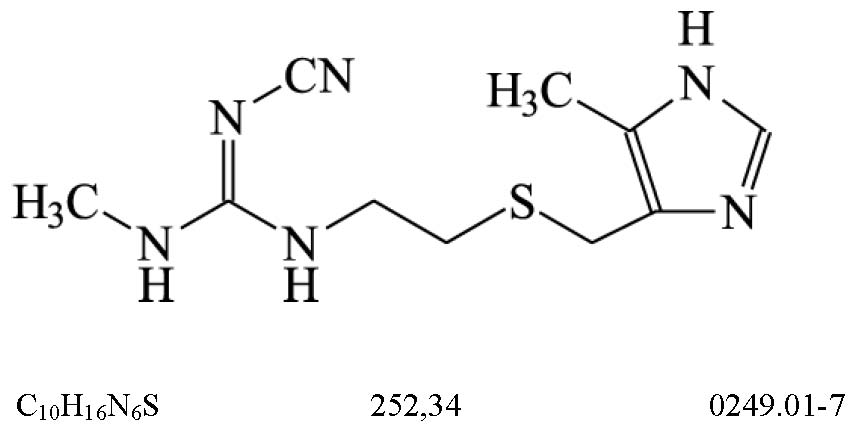
N-ciano-N'-metil-N''-{[2-(5-metil-1H-imidazol-4-il)metiltio]etil}guanidina
Contém, no mínimo, 98,5% e, no máximo, 101,5% de C10H16N6S, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó branco ou quase branco.
Solubilidade. Ligeiramente solúvel em água, solúvel etanol e praticamente insolúvel em diclorometano e em éter etílico. Solúvel emácidos minerais diluídos.
Constantes físico-químicas
Faixa de fusão (V.2.2): 139 ºC a 144 ºC. Se necessário, dissolver a substância em 2-propanol, evaporar até secura e determinar novamente a faixa de fusão.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) da amostra dessecada a 105 oC, até peso constante, e dispersa em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de cimetidina padrão, preparada de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 200 a 400 nm, de uma solução a 0,0005% (p/V) em ácido clorídrico 0,1 M exibe máximo em 221 nm, idêntico ao observado no espectro de solução similar de cimetidina padrão.
C. Proceder conforme descrito em Substâncias relacionadas. A mancha principal obtida com a solução (2) é similar em posição, cor e tamanho àquela obtida com a solução (6).
D. Dissolver cerca de 1 mg da amostra em mistura de 1 ml de etanol absoluto e 5 ml de solução de ácido cítrico a 2% (p/V) em anidrido acético, recentemente preparada. Aquecer em banho-maria durante 10 a 15 minutos. Desenvolve-se coloração violeta-avermelhada.
ENSAIOS DE PUREZA
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de amônia 13,5 M-metanol-acetato de etila (15:20:65), como fase móvel. Saturar a cuba, por 15 minutos, com o vapor da fase móvel. Aplicar, separadamente, à placa, 5 μl de cada uma das soluções descritas a seguir.
Solução (1): dissolver 0,5 g da amostra em 10 ml de metanol.
Solução (2): diluir 1 ml da solução (1) para 10 ml com metanol.
Solução (3): diluir 1 ml da solução (1) para 100 ml com metanol e diluir 20 ml desta solução para 100 ml com metanol.
Solução (4): diluir 5 ml da solução (3) para 10 ml com metanol.
Solução (5): diluir 5 ml da solução (4) para 10 ml com metanol.
Solução (6): dissolver 10 mg de cimetidina padrão em 2 ml de metanol.
Desenvolver o cromatograma. Remover a placa, secar em corrente de ar, deixar sob vapor de iodo até obter o máximo contraste das manchas e examinar sob luz ultravioleta (254 nm). Qualquer mancha secundária obtida com a solução (1) não é mais intensa que a mancha principal obtida com a solução (3) e no máximo duas manchas podem ser mais intensas que a mancha principal obtida com a solução (4).
Para que o ensaio seja válido, o cromatograma obtido com a solução (5) deve apresentar uma mancha nitidamente visível.
Metais pesados (V.3.2.3 - Método II). Ferver 1 g da amostra com 20 ml de água, por 10 minutos. Filtrar, quantitativamente, para tubo de Nessler de 50 ml e prosseguir conforme descrito em Ensaio-limite para metais pesados. No máximo 0,002% (20 ppm).
Perda por dessecação (V.2.9). Determinar, em 1 g de amostra, em estufa a 105 ºC, até peso constante. No máximo 0,5%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g da amostra. No máximo 0,2%.
DOSEAMENTO
Proceder conforme descrito em Titulação em meio não-aquoso (V.3.4.5). Dissolver 0,2 g de amostra em 60 ml de anidrido acético.
Titular com ácido perclórico 0,1 M SV, determinando o ponto final potenciometricamente. Cada ml de ácido perclórico 0,1 M SV equivale a 25,23 mg de C10H16N6S.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Anti-histamínico H2.
136.1
CIMETIDINA COMPRIMIDOS
Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C10H16N6S.
IDENTIFICAÇÃO
O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 200 a 400 nm, da solução amostra obtida no Doseamento, exibe máximo em 221 nm, idêntico ao observado no espectro da solução padrão.
CARACTERISTICAS
Teste de desintegração (V.1.4.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: água, 900 ml
Aparelhagem: cesta, 100 rpm
Tempo: 15 minutos
Procedimento: retirar alíquota do meio de dissolução, filtrar, se necessário, e diluir com ácido sulfúrico 0,1 M até concentração adequada.
Medir as absorvâncias das soluções em 218 nm (V.2.14.-3), utilizando ácido sulfúrico 0,1 M para ajuste do zero. Calcular a quantidade de cimetidina dissolvida no meio, comparando as leituras obtidas com a da solução de cimetidina padrão, na concentração de 0,0005% (p/V) preparada no mesmo solvente.
Tolerância: não menos que 75% (T) da quantidade declarada de C10H16N6S se dissolvem em 15 minutos.
DOSEAMENTO
Pesar e pulverizar 20 comprimidos. Transferir quantidade de pó equivalente a 0,1 g de cimetidina para balão volumétrico de 200 ml, adicionar 50 ml de ácido clorídrico 0,1 M. Agitar por 30 minutos e completar o volume com o mesmo solvente. Filtrar. Diluir, sucessivamente, em ácido clorídrico 0,1 M, até concentração de 0,0005% (p/V). Preparar solução padrão na mesma concentração, utilizando o mesmo solvente. Medir as absorvâncias (V.2.14.-3) das soluções resultantes em 221 nm, utilizando ácido clorídrico 0,1 M para ajuste do zero. Calcular a quantidade de C10H16N6S nos comprimidos a partir das leituras obtidas.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, à temperatura ambiente e protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
136.2
CIMETIDINA SOLUÇÃO INJETÁVEL
Cimetidina solução injetável é uma solução estéril de cloridrato de cimetidina em água para injetáveis. Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C10H16N6S.
IDENTIFICAÇÃO
A. Diluir a solução injetável, sucessivamente, em ácido clorídrico 0,1 M, de modo a obter concentração de 0,0005% (p/V) de cimetidina.
Utilizar cloridrato de cimetidina padrão e o mesmo solvente, para preparar solução na mesma concentração em cimetidina. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 200 a 400 nm, exibe máximos e mínimos somente nos mesmos comprimentos de onda observados no espectro da solução padrão.
B. A solução injetável responde às reações do íon cloreto (V.3.1.1).
CARACTERÍSTICAS
Determinação de volume (V.1.2). Cumpre o teste.
pH (V.2.19). 3,8 a 6,0.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1). Cumpre o teste. Utilizar o método de inoculação direta ou filtração por membrana.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 1 a 4 mg/kg num volume entre 0,5 e 10 ml/kg de peso do animal.
DOSEAMENTO
Transferir um volume da solução injetável, equivalente a 0,1 g de cimetidina, para balão volumétrico de 200 ml, completar o volume com ácido clorídrico 0,1 M e homogeneizar. Diluir sucessivamente com o mesmo solvente, até concentração de 0,0005% (p/V). Preparar solução padrão na mesma concentração e no mesmo solvente, utilizando cloridrato de cimetidina padrão. Medir as absorvâncias (V.2.14.-3) das soluções em 221 nm, utilizando ácido clorídrico 0,1 M como branco. Calcular a quantidade de C10H16N6S na solução injetável, a partir das leituras obtidas.
EMBALAGEM E ARMAZENAMENTO
Em recipiente de dose simples de vidro tipo I, à temperatura ambiente e protegido da luz.
ROTULAGEM
Observar a legislação vigente.
137
CIPROFLOXACINO
Ciprofloxacinum
Ácido 1-ciclopropil-6-fluor-1,4-diidro-4-oxo-7-(1-piperazinil)-3-quinolino carboxílico
Contém, no mínimo, 98,0% e, no máximo, 102,0% de C17H18FN3O3, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó cristalino ligeiramente amarelado.
Solubilidade. Praticamente insolúvel em água, muito pouco solúvel em etanol e cloreto de metileno. Solúvel em ácido acético diluído.
Constantes físico-químicas.
Faixa de fusão (V.2.2): 255 °C a 257 °C, com decomposição.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) de amostra dessecada até peso constante, e dispersa em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de ciprofloxacino padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de cloreto de metileno-metanol-hidróxido de amônio-acetonitrila (4:4:2:1), como fase móvel. Aplicar, separadamente, à placa, 5 μl de cada uma das soluções recentemente preparadas, descritas a seguir.
Solução (1): solução a 1% (p/V) de amostra em hidróxido de amônio 6 M.
Solução (2): solução a 1% (p/V) de ciprofloxacino padrão em hidróxido de amônio 6 M.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e examinar as manchas sob luz ultravioleta (254 nm e 336 nm). A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (2).
ENSAIOS DE PUREZA
Aspecto da solução. A solução a 2,5% (p/V) em ácido clorídrico 0,1 M é límpida a levemente opaca.
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de cloreto de metileno-metanol-hidróxido de amônio-acetonitrila (4:4:2:1), como fase móvel. Aplicar, separadamente,à placa, 5 μl de cada uma das soluções recentemente preparadas, descritas a seguir.
Solução (1): solução a 1% (p/V) de amostra em ácido acético 0,1 M.
Solução (2): transferir 5 mg de ácido fluoroquinolônico padrão para um balão volumétrico de 50 ml contendo 0,05 ml de hidróxido de amônio 6 M e completar o volume com água. Transferir 2 ml desta solução para balão volumétrico de 10 ml e completar o volume com água.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e xaminar sob luz ultravioleta (254 nm e 336 nm). A mancha da solução (1) de mesmo Rf da mancha principal da solução (2) não é maior em tamanho e intensidade que a mancha principal da solução (2).
Cloreto. Pesar 0,5 g de amostra, adicionar 30 ml de água, agitar por 5 minutos e filtrar através de papel filtro isento de cloro. Transferir 15 ml do filtrado para um tubo de Nessler de 50 ml. Para outro tubo de Nessler transferir 10 ml de uma solução padrão de cloreto de sódio com concentração de 8,2 μg/ml, correspondendo a 5 μg/ml de cloro, adicionar 5 ml de água e misturar. Adicionar aos tubos da amostra e do padrão, 1 ml de ácido nítrico 2 M e misturar. Adicionar, em seguida, 1 ml de nitrato de prata 0,1 M e misturar. A turbidez exibida pela preparação amostra não excede àquela produzida pela preparação padrão. No máximo 0,02% (200 ppm).
Metais pesados (V.3.2.3 - Método II). No máximo 0,002% (20 ppm).
Perda por dessecação (V.2.9). Determinar em estufa a vácuo a 120°C, por 6 horas. No máximo 1%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,1%.
DOSEAMENTO
A. Por titulação em meio não-aquoso (V.3.4.5). Dissolver 0,3 g de amostra em 80 ml de ácido acético glacial e titular com ácido perclórico 0,1 M SV. Determinar o ponto final potenciometricamente.
Cada ml de ácido perclórico 0,1 M SV equivale a 33,14 mg de C17H18FN3O3.
B. Por cromatografia líquida de alta eficiência (V.2.17.4). Utilizar cromatógrafo provido de detector ultravioleta a 278 nm, coluna de 250 mm de comprimento e 4 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octadecilsilano (3 μm a 10 μm), mantida a 30 °C; fluxo da fase móvel de 1,5 ml/minuto.
Fase móvel: mistura de ácido fosfórico 0,025 M com pH previamente
ajustado com trietilamina para 3,0±0,1 e acetonitrila (87:13).
Solução amostra: pesar, exatamente, cerca de 25 mg de amostra e
transferir para um balão volumétrico de 50 ml. Adicionar 0,2 ml de
ácido fosfórico 7% (V/V). Completar o volume com a fase móvel e
homogeneizar para obter concentração de 0,5 mg/ml.
Solução padrão: pesar, exatamente, cerca de 25 mg de ciprofloxacino padrão e transferir para um balão volumétrico de 50 ml. Adicionar 0,2 ml de ácido fosfórico 7% (V/V). Completar o volume com a fase móvel e homogeneizar para obter concentração de 0,5 mg/ml.
Solução de resolução: dissolver na solução padrão quantidade da impureza de ciprofloxacino etilenodiamina (cloridrato do ácido 1- ciclopropil-6-fluor-1,4-diidro-4-oxo-7-[2-(aminoetil)amino]-3-quinolino carboxílico) para obter solução a 0,5 mg/ml.
A eficiência da coluna não deve ser menor do que 10 000 pratos teóricos/metro, os tempos de retenção relativos são aproximadamente 0,7 para a impureza da solução de resolução e 1,0 para o ciprofloxacino.
O fator de resolução entre o pico da impureza e o do ciprofloxacino não é menor que 6. O desvio padrão relativo das áreas de replicatas dos picos registrados não é maior que 1,5%.
Procedimento: injetar separadamente 10 μl das soluções amostra e padrão, registrar os cromatogramas e medir as áreas dos picos. Calcular o teor de C17H18FN3O3 na solução amostra a partir das respostas obtidas para solução padrão e solução amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, a temperatura ambiente e protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antimicrobiano.
137.1
CIPROFLOXACINO COMPRIMIDOS
Ciprofloxacino comprimidos contêm cloridrato de ciprofloxacino equivalente a, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C17H18FN3O3.
IDENTIFICAÇÃO
A. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de cloreto de metileno-metanol-hidróxido de amônio-acetonitrila (4:4:2:1), como fase móvel. Aplicar, separadamente, à placa, 5 μl de cada uma das soluções recentemente preparadas, descritas a seguir.
Solução (1): pesar e pulverizar os comprimidos. Transferir para balão volumétrico de 100 ml, quantidade de pó equivalente a 0,15 g de ciprofloxacino. Adicionar 70 ml de água, agitar por cerca de 20 minutos e completar o volume com o mesmo solvente. Centrifugar e utilizar porção límpida do sobrenadante.
Solução (2): solução aquosa de cloridrato de ciprofloxacino padrão na concentração de 0,15% (p/V) em ciprofloxacino.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar por 15 minutos, examinar sob luz ultravioleta (254 nm e 336 nm). A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (2).
B. Proceder conforme descrito no método B do Doseamento. O tempo de retenção do pico principal obtido com a solução amostra correspondeàquele do pico principal da solução padrão.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: água, 900 ml
Aparelhagem: pás, 50 rpm
Tempo: 30 minutos
Procedimento: após o teste, retirar alíquota do meio de dissolução, filtrar e diluir em água até concentração adequada. Medir as absorvâncias das soluções em 272 nm (V.2.14.-3), utilizando água para ajuste do zero. Calcular a quantidade de C17H18FN3O3 dissolvido no meio, comparando as leituras obtidas com a da solução padrão de cloridrato de ciprofloxacino na concentração de 0,0004% em ciprofloxacino (p/V) preparada no mesmo solvente.
Tolerância: não menos que 80% (T) da quantidade declarada de C17H18FN3O3 se dissolvem em 30 minutos.
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito em Determinação da potência na monografia de cloridrato de ciprofloxacino. Preparar a solução amostra como descrito a seguir.
Solução amostra: pesar e pulverizar 20 comprimidos. Pesar, exatamente, quantidade do pó equivalente a 1,25 g de ciprofloxacino e transferir para balão volumétrico de 500 ml com auxílio de 400 ml deágua. Agitar por 30 minutos, completar o volume com água e filtrar.
Diluir, sucessivamente, até as concentrações de 4 μg/ml, 8 μg/ml e 16μg/ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) como diluente.
Solução padrão: pesar, em cloridrato de ciprofloxacino padrão, o equivalente a 20 mg de ciprofloxacino, transferir para balão volumétrico
DOSEAMENTO
A. Por espectrofotometria de absorção no ultravioleta (V.2.14.-3).
Pesar e pulverizar 20 comprimidos. Transferir para balão volumétrico de 500 ml, quantidade do pó equivalente a 1,25 g de ciprofloxacino, com auxílio de 400 ml de água. Agitar por 30 minutos, completar o volume e filtrar. Diluir, sucessivamente, até a concentração de 0,0004% (p/V), utilizando água como solvente. Utilizar cloridrato de ciprofloxacino padrão e água como solvente, para preparar solução na mesma concentração em ciprofloxacino. Medir as absorvâncias das soluções resultantes em 272 nm, utilizando água para ajuste do zero. Calcular o teor de C17H18FN3O3 nos comprimidos, a partir das leituras obtidas.
B. Por cromatografia líquida de alta eficiência (V.2.17.4). Utilizar cromatógrafo provido de detector ultravioleta a 278 nm; coluna de 250 mm de comprimento e 4 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octadecilsilano (3 μm a 10 μm), mantida a 30 °C; fluxo da fase móvel de 1,5 ml/minuto.
Fase móvel: mistura de ácido fosfórico 0,025 M com pH ajustado previamente com trietilamina para 3,0 ± 0,1 e acetonitrila (87:13).
Solução amostra: pesar e pulverizar 20 comprimidos. Transferir quantidade do pó equivalente a 1,25 g de ciprofloxacino para balão volumétrico de 500 ml, com auxílio de 400 ml de água. Agitar por 30 minutos, completar o volume com água e filtrar. Diluir, sucessivamente, até concentração de 0,25 mg/ml, utilizando água como solvente.
Solução padrão: pesar, exatamente, cerca de 30 mg de cloridrato de ciprofloxacino padrão, transferir para balão volumétrico de 100 ml e completar o volume com água de modo a obter solução a 0,25 mg/ml de ciprofloxacino.
Solução de resolução: dissolver na solução padrão quantidade da impureza de ciprofloxacino etilenodiamina (cloridrato do ácido 1- ciclopropil-6-fluor-1,4-diidro-4-oxo-7-[2-(aminoetil)amino]-3-quinolino carboxílico) para obter solução a 0,5 mg/ml.
A eficiência da coluna não deve ser menor do que 10 000 pratos teóricos/metro, os tempos de retenção relativos são aproximadamente 0,7 para a impureza da solução de resolução e 1,0 para o ciprofloxacino.
O fator de resolução entre o pico da impureza e o do ciprofloxacino não é menor que 6. O desvio padrão relativo das áreas de replicatas dos picos registrados não é maior que 1,5%.
Procedimento: injetar, separadamente, 20 μl das soluções padrão e amostra, registrar os cromatogramas e medir as áreas dos picos.
Calcular a quantidade de C17H18FN3O3 na solução amostra a partir das respostas obtidas para solução padrão e solução amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
137.2
CIPROFLOXACINO SOLUÇÃO INJETÁVEL
Ciprofloxacino solução injetável é uma solução estéril de ciprofloxacino em água para injetáveis, em solução de glicose a 5% (p/V) ou de cloreto de sódio a 0,9% (p/V), preparada com a ajuda de ácido lático. Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C17H18FN3O3.
IDENTIFICAÇÃO
Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de cloreto de metileno-metanol-hidróxido de amônio-acetonitrila (4:4:2:1), como fase móvel. Aplicar, separadamente, à placa, previamente saturada por 15 minutos em atmosfera de amônia, 10 μl de cada uma das soluções recentemente preparadas, descritas a seguir.
Solução (1): diluir volume da solução injetável em água de forma a obter concentração de cerca de 0,05% (p/V) de ciprofloxacino.
Solução (2): solução de cloridrato de ciprofloxacino padrão em água, na concentração de 0,05% (p/V) de ciprofloxacino.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e examinar sob luz ultravioleta (254 nm e 336 nm). A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidadeàquela obtida com a solução (2).
CARACTERÍSTICAS
Determinação de volume (V.1.2). Cumpre o teste.
pH (V.2.19). 3,5 a 4,6.
ENSAIOS DE PUREZA
Limite da impureza ciprofloxacino etilenodiamina. Proceder conforme descrito no método B de Doseamento. Calcular a porcentagem da impureza, a partir do cromatograma obtido com a solução amostra, pela expressão: 100 [0,7 Ri / (0,7 Ri + Rc)]...., onde 0,7 é o fator de resposta entre a impureza e o ciprofloxacino, Ri é a resposta do pico da impureza e Rc é a resposta do pico de ciprofloxacino. No máximo 0,5%.
Conteúdo de ácido lático. Proceder conforme descrito em Cromatografia líquida de alta eficiência (V.2.17.4), utilizando cromatógrafo líquido equipado com detector ultravioleta a 208 nm, coluna de 300 mm de comprimento e 7,8 mm de diâmetro interno, empacotada com resina de troca iônica constituída de copolímero de estireno-divinilbenzeno sulfonatado na forma hidrogenada (7 a 11 μm), mantida a 40 °C; fluxo da fase móvel de 0,6 ml/minuto.
Fase móvel: mistura de ácido sulfúrico 0,0025 M e acetonitrila (85:15).
Solução amostra: utilizar a solução injetável não diluída.
Solução padrão: solução a 0,8 mg/ml de lactato de sódio padrão em água.
A eficiência da coluna não deve ser menor que 5 000 pratos teóricos/metro. O desvio padrão relativo das áreas de replicatas dos picos registrados não deve ser maior que 2,0%.
Procedimento: injetar, separadamente, 20 μl das soluções amostra e padrão, registrar os cromatogramas e medir as áreas dos picos. Calcular a quantidade, em mg, de C3H6O3 por ml da solução injetável pela expressão: (90,08/112,07) (C) (Ra/Rp), em que 90,08 e 112,07 são as massas moleculares do ácido láctico e do lactato de sódio, respectivamente, C é a concentração, em mg/ml, de padrão de lactato de sódio, e Ra e Rp são as respostas dos picos obtidos com a solução amostra e padrão, respectivamente. O valor obtido está entre 0,288 e 0,325 mg de C3H6O3 por mg de ciprofloxacino rotulado.
Conteúdo de dextrose (se presente). Transferir 50 ml de solução injetável para balão volumétrico de 100 ml. Adicionar 0,2 ml de hidróxido de amônio 6 M, completar o volume com água e agitar.
Determinar o ângulo de rotação (α), em tubo de 200 mm a 25 oC (V.2.8). A quantidade, em g, de C6H12O6.H2O em 100 ml de solução injetável é calculada pela expressão: 2 (1,0425 α). O valor obtido está entre 4,75 e 5,25 g de C6H12O6.H2O por 100 ml de solução injetável.
Conteúdo de cloreto de sódio (se presente). Transferir 10 ml da solução injetável para um erlenmeyer, diluir com água até aproximadamente 150 ml, adicionar 1,5 ml de cromato de potássio SI, e titular com nitrato de prata 0,1 M SV. Cada ml de nitrato de prata equivale a 5,844 mg de cloreto de sódio. O valor obtido está entre 85,5 mg e 94,5 mg.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1). Cumpre o teste. Usar o método de filtração por membrana.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 1 ml/kg, de uma solução contendo 20 mg/ml de ciprofloxacino, em água para injetáveis.
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito em Determinação da potência na monografia de cloridrato de ciprofloxacino. Preparar a solução amostra e solução podrão como descrito a seguir.
Solução amostra: transferir volume da solução injetável contendo o equivalente a 1,25 g de ciprofloxacino para balão volumétrico de 500 ml, com auxílio de 400 ml de água, homogeneizar e completar o volume com o mesmo diluente. Diluir, sucessivamente, até as concentrações de 4 μg/ml, 8 μg/ml e 16 μg/ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) como diluente.
Solução padrão: pesar, em cloridrato de ciprofloxacino padrão, o equivalente a 20 mg de ciprofloxacino, transferir para balão volumétrico de 50 ml e completar o volume com água. Diluir, sucessivamente, até as concentrações de 4 μg/ml, 8 μg/ml e 16 μg/ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) como diluente.
DOSEAMENTO
A. Por espectrofotometria de absorção no ultravioleta (V.2.14.-3). Diluir a solução injetável, em água, de modo a obter concentração de cerca de 0,0004% (p/V) de ciprofloxacino. Utilizar cloridrato de ciprofloxacino padrão e água como solvente, para preparar solução na mesma concentração em ciprofloxacino. As soluções devem ser mantidas ao abrigo da luz. Medir as absorvâncias das soluções em 272 nm, utilizando água para o ajuste do zero. Calcular a quantidade de C17H18FN3O3 na solução injetável, a partir das leituras obtidas.
B. Por cromatografia líquida de alta eficiência (V.2.17.4). Utilizar cromatógrafo provido de detector ultravioleta a 278 nm; coluna de 250 mm de comprimento e 4,0 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octadecilsilano (3 μm a 10 μm), mantida a 30 °C; fluxo da fase móvel de 1,5 ml/minuto.
Fase móvel: mistura de ácido fosfórico 0,025 M com pH previamente ajustado com trietilamina para 3,0 ± 0,1 e acetonitrila (87:13).
Solução amostra: transferir volume da solução injetável equivalente a 25 mg de ciprofloxacino para balão volumétrico de 100 ml e completar o volume com a fase móvel.
Solução padrão: pesar, exatamente, cerca de 30 mg de cloridrato de ciprofloxacino padrão e transferir para balão volumétrico de 100 ml, utilizando fase móvel como solvente, para obter uma concentração de 0,25 mg/ml de ciprofloxacino.
Solução de resolução: dissolver na solução padrão quantidade da impureza ciprofloxacino etilenodiamina (cloridrato do ácido 1-ciclopropil- 6-fluor-1,4-diidro-4-oxo-7-[2-(aminoetil)amino]-3-quinolino carboxílico) para obter solução a 0,25 mg/ml.
A eficiência da coluna não deve ser menor do que 10 000 pratos teóricos/metro, os tempos de retenção relativos são aproximadamente 0,7 para a impureza da solução de resolução e 1,0 para o ciprofloxacino.
O fator de resolução entre o pico da impureza e o do ciprofloxacino não é menor que 6. O desvio padrão relativo das áreas de replicatas dos picos registrados não é maior que 1,5%.
Procedimento: injetar, separadamente, 10 μl das soluções padrão e amostra, registrar os cromatogramas e medir as áreas dos picos.
Calcular a quantidade de C17H18FN3O3 na solução amostra a partir das respostas obtidas para solução padrão e solução amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes de vidro tipo I, em local fresco e protegidos da luz.
Evitar congelamento.
ROTULAGEM
Observar a legislação vigente.
137.3
CIPROFLOXACINO SOLUÇÃO OFTÁLMICA
Ciprofloxacino solução oftálmica é uma solução aquosa, estéril de cloridrato de ciprofloxacino. Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C17H18FN3O3.
IDENTIFICAÇÃO
Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de cloreto de metileno-metanol-hidróxido de amônio-acetonitrila (4:4:2:1), como fase móvel. Aplicar, separadamente, à placa, 3 μl de cada uma das soluções recentemente preparadas, descritas a seguir.
Solução (1): diluir volume da solução oftálmica em água, para obter solução a 0,3% (p/V) de ciprofloxacino.
Solução (2): solução a 0,35% (p/V) de cloridrato de ciprofloxacino padrão em água.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar por 15 minutos, examinar sob luz ultravioleta (254 nm e 336 nm). A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (2).
CARACTERÍSTICAS
Determinação de volume (V.1.2). Cumpre o teste.
pH (V.2.19). 3,5 a 5,5.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1). Cumpre o teste. Usar o método de filtração por membrana.
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito em Determinação da potência na monografia de cloridrato de ciprofloxacino, preparando a solução amostra e solução padrão como descrito a seguir.
Solução amostra: utilizar volume da solução oftálmica correspondente a 40 mg de ciprofloxacino, transferir para balão volumétrico de 100 ml e completar o volume com água. Diluir, sucessivamente, até as concentrações de 4 μg/ml, 8 μg/ml e 16 μg/ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) como diluente.
Solução padrão: pesar, em cloridrato de ciprofloxacino padrão, o equivalente a 20 mg de ciprofloxacino, transferir para balão volumétrico de 50 ml e completar o volume com água. Diluir, sucessivamente, até as concentrações de 4 μg/ml, 8 μg/ml e 16 μg/ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) como diluente.
DOSEAMENTO
Procederconforme descrito em Cromatografia líquida de alta eficiência (V.2.17.4), utilizando cromatógrafo provido de detector ultravioleta a 280 nm; coluna de 250 mm de comprimento e 4,6 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octadecilsilano (3 μm a 10 μm), mantida a 30 °C; fluxo da fase móvel 1,5 ml/minuto.
Fase móvel: mistura de fosfato de tetrabutilamônio 0,005 M com pH
ajustado previamente com ácido fosfórico para 2,0 e metanol
(75:25).
Solução amostra: utilizar volume da solução oftálmica correspondente a 6 mg de ciprofloxacino, transferir para balão volumétrico de 50 ml e completar o volume com água para obter concentração de 0,12 mg/ml de ciprofloxacino.
Solução padrão: pesar, exatamente, quantidade de cloridrato de ciprofloxacino padrão e diluir em água para obter concentração de 0,12 mg/ml de ciprofloacino.
Solução de resolução: dissolver na solução padrão uma quantidade da impureza ciprofloxacino etilenodiamina (cloridrato do ácido 1-ciclopropil- 6-fluor-1,4-diidro-4-oxo-7-[2-(aminoetil)amino]-3-quinolino carboxílico) para obter solução a 0,01 mg/ml.
A eficiência da coluna não deve ser menor que 2 000 pratos teóricos/metro. Os tempos de retenção relativos são cerca de 0,8 para a impureza da solução de resolução e de 1,0 para o ciprofloxacino. A resolução entre o pico da impureza e o pico do ciprofloxacino não é menor do que 1,5. O desvio padrão relativo das áreas de replicatas dos picos registrados não é maior que 2,0%.
Procedimento: injetar, separadamente, 20 μl das soluções padrão e amostra, registrar os cromatogramas e medir as áreas dos picos.
Calcular a quantidade de C17H18FN3O3 da solução amostra a partir dos picos obtidos com as soluções amostra e padrão.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
138
CLORETO DE POTÁSSIO
Kalii chloridum
KCl 74,55 1023.05-5
Contém, no mínimo, 99,0% e, no máximo, 100,5% de KCl, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó branco cristalino ou cristais incolores.
Solubilidade. Muito solúvel em água, praticamente insolúvel em etanol.
IDENTIFICAÇÃO
A. Responde às reações do íon cloreto (V.3.1.1).
B. Responde às reações do íon potássio (V.3.1.1).
ENSAIOS DE PUREZA
Aspecto da solução. A solução de cloreto de potássio a 10% (p/V) emágua isenta de dióxido de carbono é límpida e incolor.
Acidez ou alcalinidade. Adicionar 0,1 ml de azul de bromotimol SI a 50 ml da solução obtida em Aspecto da solução. Não é necessário mais que 0,5 ml de solução de ácido clorídrico 0,01 M SV ou de solução de hidróxido de sódio 0,01 M SV para promover a viragem do indicador.
Perda por dessecação: (V.2.9). Determinar em 1 g de amostra, em estufa entre 100 °C e 105 °C, por 3 horas. No máximo 1%.
Brometos. Diluir 1 ml da solução obtida em Aspecto da solução para 50 ml com água. A 5 ml desta solução, adicionar 2 ml de vermelho de fenol SR, 1 ml de cloramina 0,01% (p/V) em água, recentemente preparada e homogeneizar. Após 2 minutos adicionar 0,15 ml de solução de tiossulfato de sódio 0,1 M, misturar e diluir com água para 10 ml. A absorvância (V.2.14) da solução, medida em 590 nm, utilizandoágua para o ajuste do zero não é maior do que a da solução padrão, preparada do mesmo modo e, ao mesmo tempo, utilizando 5 ml de solução de brometo de potássio a 0,3% (p/V). No máximo 0,1%.
Iodetos. Umedecer 5 g da amostra pela adição, gota a gota, da mistura recém-preparada de 0,15 ml da solução de nitrito de sódio 10% (p/V), 2 ml de ácido sulfúrico 0,5 M, 25 ml de amido SI e 25 ml de água. Examinar à luz do dia após 5 minutos. Não se desenvolve coloração azul.
Sulfato (V.3.2.2). Utilizar 5 ml da solução obtida em Aspecto da solução e prosseguir conforme descrito em Ensaio-limite para sulfatos. No máximo 0,03% (300 ppm).
Bário. A 5 ml da solução obtida em Aspecto da solução, adicionar 5 ml de água e 1 ml de ácido sulfúrico 5,5% (V/V). Após 15 minutos qualquer opalescência observada não é mais intensa do que a da mistura de 5 ml da solução obtida em Aspecto da solução e 6 ml deágua.
Metais pesados (V.3.2.3 - Método II). Utilizar 10 ml da solução
obtida em Aspecto da solução e prosseguir conforme descrito em
Ensaio-limite para metais pesados. No máximo 0,001% (10 ppm).
Ferro (V.3.2.4). Utilizar 5 ml da solução obtida em Aspecto da solução e prosseguir conforme descrito em Ensaio-limite para ferro. No máximo 0,002% (20 ppm).
Magnésio e metais alcalino-terrosos (V.3.2.9). Utilizar 10 g da amostra e prosseguir conforme descrito em Ensaio-limite para magnésio e metais alcalino-terrosos. O volume de edetato de sódio 0,01 M SV utilizado não deve exceder 5 ml. No máximo 0,02% (200 ppm), calculados como cálcio.
Sódio. Exigido para cloreto de potássio destinado à preparação de soluções para uso parenteral ou soluções para hemodiálise. Proceder conforme descrito em Espectrofotometria de emissão atômica (V.2.23). Utilizar as soluções descritas a seguir e medir as intensidades de emissão em 589 nm. No máximo 0,1% (1 000 ppm).
Solução amostra: dissolver 1 g da amostra em água e diluir para 100 ml com o mesmo solvente.
Solução referência: dissolver em água destilada 0,5084 g de cloreto de sódio, previamente dessecado entre 100 oC e 105 oC, por 3 horas e diluir para 1 000 ml com o mesmo solvente (0,2 mg de sódio por ml). Diluir se necessário.
Alumínio (V.3.2.10). Exigido para cloreto de potássio destinado à preparação de soluções para hemodiálise. Dissolver 4 g da amostra em 100 ml de água e adicionar 10 ml de solução tampão acetato pH 6,0. Prosseguir conforme descrito em Ensaio-limite para alumínio.
Utilizar como solução de referência mistura de 2 ml de solução padrão de alumínio (2 ppm), 10 ml de solução tampão acetato pH 6,0 e 98 ml de água. Para o branco utilizar mistura de 10 ml da solução tampão acetato pH 6,0 e 100 ml de água. No máximo 0,0001% (1 ppm).
DOSEAMENTO
Dissolver 1,3 g em água destilada e diluir para 100 ml com o mesmo solvente. A 10 ml desta solução adicionar 50 ml de água, 5 ml deácido nítrico a 20% (p/V), 25 ml da solução de nitrato de prata 0,1 M SV e 2 ml de ftalato de dibutila e homogeneizar. Titular com tiocianato de amônio 0,1 M SV, utilizando 2 ml de solução de sulfato férrico amoniacal a 10% (p/V) como indicador, agitando vigorosamente, até a viragem do indicador. Cada ml de nitrato de prata 0,1 M SV equivale a 7,46 mg de KCl.
EMBALAGEM E ACONDICIONAMENTO
Em recipientes bem-fechados.
ROTULAGEM
O rótulo do recipiente deve indicar se a substância pode ser utilizada na preparação de soluções para uso parenteral ou para hemodiálise.
CLASSE TERAPÊUTICA
Repositor eletrolítico.
________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Vermelho de fenol SR
Preparação - Adicionar 25 ml da solução A à solução B. Se necessário, ajustar pH da mistura para 4,7.
Solução A: dissolver 33 mg de vermelho de fenol em 1,5 ml de hidróxido de sódio a 8,5% (p/V) e diluir para 100 ml com água.
Solução B: dissolver 25 mg de sulfato de amônio em 235 ml de água, adicionar 105 ml de hidróxido de sódio a 8,5% (p/V) e 135 ml deácido acético a 12% (p/V).
Azul de bromotimol SI
Preparação - Dissolver 50 mg de azul de bromotimol em mistura de 4 ml de hidróxido de sódio 0,02 M e 20 ml de etanol e completar o volume para 100 ml com água.
139
CLORETO DE SÓDIO
Natrii chloridum
NaCl 58,44 1113.07- 0
Contém, no mínimo, 99,0% e, no máximo, 100,5% de NaCl, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó cristalino branco ou cristais incolores.
Solubilidade. Muito solúvel em água, praticamente insolúvel em etanol.
IDENTIFICAÇÃO
A. Responde às reações do íon cloreto (V.3.1.1).
B. Responde às reações do íon sódio (V.3.1.1).
ENSAIOS DE PUREZA
Aspecto da solução. A solução a 20% (p/V) em água isenta de dióxido de carbono é límpida e incolor.
Acidez ou alcalinidade. A 20 ml da solução descrita em Aspecto da solução, adicionar 0,1 ml de azul de bromotimol SI. Não é necessário mais que 0,5 ml de ácido clorídrico 0,01 M SV ou de hidróxido de sódio 0,01 M para promover a viragem do indicador.
Perda por dessecação (V.2.9). Determinar em 1 g de amostra, em estufa entre 100 ºC e 105 oC, por 3 horas. No máximo 1%.
Brometos. A 1 ml da solução descrita em Aspecto da solução, adicionar 4 ml de água, 2 ml de vermelho de fenol SR e 1 ml de cloramina 0,01% (p/V), recentemente preparada. Homogeneizar imediatamente.Após 2 minutos adicionar 0,15 ml de solução de tiossulfato de sódio 0,1 M, misturar e diluir para 10 ml com água. A absorvância (V.2.14) desta solução, medida em 590 nm, utilizandoágua para o ajuste do zero, não é maior do que a da solução padrão, preparada ao mesmo tempo e da mesma maneira, utilizando 5 ml de brometo de potássio a 0,3% (p/V). No máximo 0,005% (50 ppm).
Ferrocianetos. Dissolver 2 g da amostra em 6 ml de água. Adicionar 0,5 ml de mistura de 5 ml de solução a 1% (p/V) de sulfato ferroso amoniacal em solução a 0,25 % (p/V) de ácido sulfúrico e 95 ml de sulfato ferroso a 1% (p/V). Não se desenvolve coloração azul.
Iodetos. Umedecer 5 g da amostra pela adição, gota a gota, de mistura recém-preparada de 0,15 ml de nitrito de sódio 10% (p/V), 2 ml deácido sulfúrico 0,5 M, 25 ml de amido SI e 25 ml de água. Examinarà luz do dia após 5 minutos. Não se desenvolve coloração azul.
Fosfatos (V.3.2.11). Diluir 2 ml da solução descrita em Aspecto da solução para 100 ml com água e prosseguir conforme descrito em Ensaio-limite para fosfatos. No máximo 0,0025% (25 ppm).
Sulfato (V.3.2.2). Utilizar 3 ml da solução obtida em Aspecto da solução e prosseguir conforme descrito em Ensaio-limite para sulfatos. No máximo 0,025% (250 ppm).
Arsênio (V.3.2.5). Utilizar 5 ml da solução obtida em Aspecto da solução e prosseguir conforme descrito em Ensaio-limite para arsênio. No máximo 0,0001% (1 ppm).
Bário. A 2,5 ml da solução obtida em Aspecto da solução, adicionar 7,5 ml de água e 1 ml de ácido sulfúrico 5,5% (V/V). Após 15 minutos qualquer opalescência observada não é mais intensa do que a da mistura de 2,5 ml da solução obtida em Aspecto da solução e 8,5 ml de água.
Metais pesados (V.3.2.3 - Método II). Diluir 10 ml da solução descrita em Aspecto da solução para 20 ml com água. Utilizar 10 ml desta solução e prosseguir conforme descrito em Ensaio-limite para metais pesados. No máximo 0,001% (10 ppm).
Ferro (V.3.2.4). Utilizar 2,5 ml da solução obtida em Aspecto da solução e prosseguir conforme descrito em Ensaio-limite para ferro. No máximo 0,002% (20 ppm).
Magnésio e metais alcalino-terrosos (V.3.2.9). Utilizar 10 g da amostra e prosseguir conforme descrito em Ensaio-limite para magnésio e metais alcalino-terrosos. O volume de edetato de sódio 0,01 M SV utilizado não deve exceder 2,5 ml. No máximo 0,01% (100 ppm), calculados comocálcio
Potássio. Exigido para cloreto de sódio destinado à preparação de soluções para uso parenteral ou soluções para hemodiálise. Proceder conforme descrito em Espectrofotometria de emissão atômica (V.2.23 - Método I). Utilizar as soluções descritas a seguir e medir as intensidades de emissão em 768 nm. No máximo 0,05% (500 ppm)
Solução amostra: dissolver 1 g da amostra em água e diluir para 100 ml com o mesmo solvente.
Solução referência: dissolver em água destilada 1,144 g de cloreto de potássio, previamente dessecado entre 100 oC e 105 oC, por 3 horas e diluir para 1 000 ml com o mesmo solvente (600 μg de potássio por ml). Diluir se necessário.
Alumínio (V.3.2.10). Exigido para cloreto de sódio destinado à preparação de soluções para hemodiálise. Dissolver 20 g da amostra em 100 ml de água e adicionar 10 ml de solução tampão acetato pH 6,0.
Prosseguir conforme descrito em Ensaio-limite para alumínio. Utilizar como solução de referência mistura de 2 ml da solução padrão de alumínio (2 ppm), 10 ml de solução tampão de acetato pH 6,0 e 98 ml de água. Para o branco utilizar mistura de 10 ml de solução tampão acetato pH 6,0 e 100 ml de água. No máximo 0,00002% (0,2 ppm).
DOSEAMENTO
Dissolver 1 g em água e diluir para 100 ml com o mesmo solvente.A 10 ml desta solução adicionar 50 ml de água, 5 ml de ácido nítrico a 20% (p/V), 25 ml de nitrato de prata 0,1 M SV e 2 ml de ftalato de dibutila, homogeneizar. Titular com tiocianato de amônio 0,1 M SV, utilizando 2 ml da solução de sulfato férrico amoniacal a 10% (p/V) como indicador agitando, vigorosamente, até a viragem do indicador. Cada ml de nitrato de prata 0,1 M SV equivale a 5,844 mg de NaCl.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
O rótulo do recipiente deve indicar se a substância pode ser utilizada na preparação de soluções para uso parenteral ou para hemodiálise.
CLASSE TERAPÊUTICA
Repositor eletrolítico.
_________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Vermelho de fenol SR
Preparação - Adicionar 25 ml da solução A à solução B. Se necessário, ajustar pH da mistura para 4,7.
Solução A: dissolver 33 mg de vermelho de fenol em 1,5 ml de hidróxido de sódio a 8,5% (p/V) e diluir para 100 ml com água.
Solução B: dissolver 25 mg de sulfato de amônio em 235 ml de água, adicionar 105 ml de hidróxido de sódio a 8,5% (p/V) e 135 ml deácido acético a 12% (p/V).
Azul de bromotimol SI
Preparação - Dissolver 50 mg de azul de bromotimol em mistura de 4 ml de hidróxido de sódio 0,02 M e 20 ml de etanol e completar o volume para 100 ml com água.
140
CLORIDRATO DE BIPERIDENO
Biperideni hidrochloridum
Cada ml de nitrato de prata 0,1 M SV equivale a 5,844 mg de NaCl.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
O rótulo do recipiente deve indicar se a substância pode ser utilizada na preparação de soluções para uso parenteral ou para hemodiálise.
CLASSE TERAPÊUTICA
Repositor eletrolítico.
_________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Vermelho de fenol SR
Preparação - Adicionar 25 ml da solução A à solução B. Se necessário, ajustar pH da mistura para 4,7.
Solução A: dissolver 33 mg de vermelho de fenol em 1,5 ml de hidróxido de sódio a 8,5% (p/V) e diluir para 100 ml com água.
Solução B: dissolver 25 mg de sulfato de amônio em 235 ml de água, adicionar 105 ml de hidróxido de sódio a 8,5% (p/V) e 135 ml deácido acético a 12% (p/V).
Azul de bromotimol SI
Preparação - Dissolver 50 mg de azul de bromotimol em mistura de 4 ml de hidróxido de sódio 0,02 M e 20 ml de etanol e completar o volume para 100 ml com água.
140
CLORIDRATO DE BIPERIDENO
Biperideni hidrochloridum
Cloridrato de α-biciclo[2.2.1]hepta-5-eno-2-il-α-fenil-1-piperidinopropanol
Contém, no mínimo, 98,0% e, no máximo, 101,0% de C21H29NO.HCl, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó cristalino, branco e praticamente inodoro.
Solubilidade. Ligeiramente solúvel em água, pouco solúvel em metanol, ligeiramente solúvel em éter etílico, etanol e clorofórmio.
Constantes físico-químicas
Ponto de fusão (V.2.2): funde a 275 °C, com decomposição.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) da amostra dessecada e dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro do cloridrato de biperideno padrão.
B. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 200 nm a 400 nm, de uma solução 0,1% (p/V) em metanol, exibe máximos e mínimos somente nos mesmos comprimentos de onda de solução similar de cloridrato de biperideno padrão. As respectivas absorvâncias, calculadas em relação à substância dessecada, no comprimento de onda de absorvância máxima, em cerca de 275 nm, não diferem mais que 3%.
C. Dissolver cerca de 20 mg em 5 ml de ácido fosfórico. Produz-se coloração verde.
D. Uma alíquota de 5 ml de solução a 0,2% (p/V) responde às reações do íon cloreto (V.3.1.1).
ENSAIOS DE PUREZA
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel G, como suporte, e mistura de tolueno-metanol-ácido acético glacial (45:8:4), como fase móvel. Aplicar, separadamente, à placa, 20 μl de cada uma das soluções descritas a seguir, preparadas em etanol-água (1:1).
Solução (1): solução a 0,25% (p/V) da amostra.
Solução (2): solução a 0,005% (p/V) de cloridrato de biperideno padrão.
Desenvolver o cromatograma. Remover a placa e deixar secar ao ar.
Expor a placa por 10 minutos a vapores de iodo em cuba fechada previamente equilibrada, tendo ao fundo cristais de iodo.
Nenhuma mancha secundária obtida no cromatograma com a solução (1) é mais intensa que a mancha obtida com a solução (2).
Perda por dessecação (V.2.9). Determinar em 1 g de amostra, em estufa a 105° C, por 3 horas. No máximo 0,5%.
DOSEAMENTO
Proceder conforme descrito em titulação em meio não-aquoso (V.3.4.5). Pesar, exatamente, cerca de 0,5 g de amostra e dissolver em 80 ml de ácido acético glacial, aquecendo ligeiramente, se necessário.
Esfriar, adicionar 1 gota de cloreto de metilrosanilínio SI (cristal violeta) e 10 ml de acetato de mercúrio a 6% (p/V) em ácido acético glacial. Titular com solução de ácido perclórico 0,1 M SV até viragem para azul. Realizar ensaio em branco e fazer as correções necessárias. Cada ml de ácido perclórico 0,1 M SV equivale a 34,793 mg de C21H29NO.HCl.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antiparkinsoniano, anticolinérgico.
140.1
CLORIDRATO DE BIPERIDENO COMPRIMIDOS
Contêm, no mínimo, 93,0% e, no máximo, 107,0% da quantidade declarada de C21H29NO.HCl.
IDENTIFICAÇÃO
A. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.l) utilizando sílica-gel GF254, como suporte e mistura de álcool isopropílico-hidróxido de amônio 25% (V/V) (95:5), como fase móvel. Aplicar separadamente, à placa, 20 μl de cada uma das soluções descritas a seguir.
Solução (1): pulverizar os comprimidos, pesar do pó o equivalente a 10 mg de cloridrato de biperideno, adicionar 5 ml de cloreto de metileno e agitar. Filtrar e lavar o resíduo com 5 ml de cloreto de metileno. Evaporar o filtrado. Dissolver o resíduo com 1 ml de metanol.
Solução (2): solução a 1% (p/V) do padrão em metanol.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e examinar sob luz ultravioleta (254 nm). Expor a placa por 10 minutos a vapores de iodo em cuba fechada, previamente saturada, tendo ao fundo cristais de iodo. A mancha principal obtida com a solução (1) corresponde em posição, cor e intesidade àquela obtida com a solução (2).
B. Pesar e pulverizar os comprimidos. Utilizar quantidade do pó equivalente a 10 mg de cloridrato de biperideno, adicionar 10 ml deágua e aquecer por 15 minutos. Filtrar. O filtrado responde às reações do íon cloreto (V.3.1.1).
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). No máximo 15 minutos.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
]Meio de dissolução: ácido clorídrico 0,1 M, 500 ml
Aparelhagem: pás, 50 rpm
Tempo: 45 minutos
Solução padrão: dissolver quantidade, exatamente pesada de cloridrato de biperideno padrão em metanol e diluir com o mesmo solvente de modo a obter concentração de cerca de 0,8 mg/ml. Pipetar 10 ml desta solução para balão volumétrico de 100 ml e completar o volume com água. Pipetar 5 ml da última solução para balão volumétrico de 100 ml e completar o volume com ácido clorídrico 0,1 M.
Solução amostra: após realização do teste, utilizar porções filtradas do meio de dissolução.
Branco: utilizar ácido clorídrico 0,1 M.
Procedimento: determinar, previamente, utilizando 25 ml de cada uma das soluções, (padrão, amostra e branco) o volume de hidróxido de sódio 0,1 M necessário para ajustar potenciometricamente o pH para 5,3. Transferir, separadamente, para funis de separação, 25 ml das soluções padrão, amostra e branco. Adicionar, respectivamente, a cada funil, o volume de hidróxido de sódio 0,1 M determinado anteriormente no ajuste de pH. Adicionar 5 ml da solução tampão fosfato-púrpura de bromocresol SR. Extrair com 15 ml de clorofórmio, exatamente medidos, agitando vigorosamente por 4 minutos.
Recolher o extrato clorofórmico, filtrando em papel de filtração lenta.
Medir as absorvâncias das soluções em 408 nm (V.2.14.-3), utilizando a preparação branco para ajuste do zero. Calcular o conteúdo de C21H29NO.HCl dissolvido no meio, comparando as leituras obtidas com a da preparação padrão.
Tolerância: não menos que 75% (T) da quantidade declarada de C21H29NO.HCl se dissolvem em 45 minutos.
ROTULAGEM
Observar a legislação vigente.
________________________________________________________
XII.2 REAGENTES E SOLUÇÕES REAGENTES
Solução tampão fosfato-púrpura de bromocresol SR
Preparação - Dissolver 38 g de fosfato de sódio monobásico e 2 g de fosfato de sódio dibásico anidro em água, em balão volumétrico de 1000 ml, completar o volume com água e homogeneizar. Ajustar o pH da solução para 5,3 ± 0,1, com hidróxido de sódio ou ácido fosfórico, se necessário (solução A). Dissolver 400 mg de púrpura de bromocresol em 30 ml de água, adicionar 6,3 ml de hidróxido de sódio 0,1 M e diluir com água para 500 ml (solução B). No dia de sua utilização adicionar a um funil de separação iguais volumes da solução A, solução B e clorofórmio. Agitar e desprezar a fase orgânica. Repetir a extração com igual volume de clorofórmio até que a camada orgânica se apresente incolor. Utilizar a fase aquosa.
141
CLORIDRATO DE CIPROFLOXACINO
Ciprofloxacini hydrochloridum
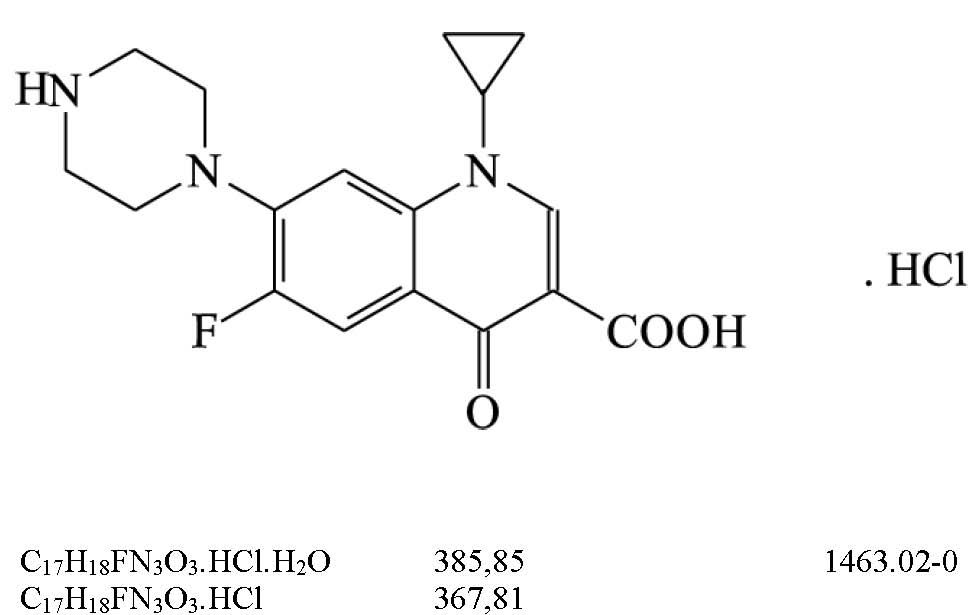
Cloridrato do ácido 1-ciclopropil-6-fluor-1,4-diidro-4-oxo-7-(1-piperazinil)- 3-quinolino carboxílico
Contém, no mínimo, 98,0% e, no máximo, 102,0% de C17H18FN3O3.HCl, em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Pó cristalino amarelo pálido.
Solubilidade. Solúvel em água, levemente solúvel em metanol e ácido acético glacial, muito pouco solúvel em etanol, praticamente insolúvel em acetona, acetato de etila e cloreto de metileno.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) da amostra dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de cloridrato de ciprofloxacino padrão, preparado de maneira idêntica.
B. 0,1 g da amostra responde à reação (2) do íon cloreto (V.3.1.1).
ENSAIOS DE PUREZA
pH (V.2.19). 3,0 a 4,5. Determinar em solução a 2,5% (p/V) em água livre de dióxido de carbono.
Água (V.2.20.1). Entre 4,7% e 6,7%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,1%.
Metais pesados (V.3.2.3 - Método II). No máximo 0,002% (20 ppm).
Impurezas inorgânicas (V.3.2). Cumpre o teste.
Ácido fluorquinolônico. Proceder conforme descrito em Cromatografia em camada delgada utilizando sílica-gel GF254 como suporte, e mistura de cloreto de metileno-metanol-hidróxido de amônio-acetonitrila (4:4:2:1), como fase móvel. Saturar a cuba com hidróxido de amônio por 15 minutos. Aplicar, separadamente, à placa, 5 μl de cada uma das seguintes soluções recentemente preparadas, descritas a seguir.
Solução (1): solução a 1% (p/V) de amostra em água.
Solução (2): solução a 0,01% (p/V) de cloridrato de ciprofloxacino padrão contendo 0,05 ml de hidróxido de amônio 6 M em água.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e examinar sob luz ultravioleta (254 nm e 336 nm). A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidadeàquela obtida com a solução (2).
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17), pelo método de difusão em agar, utilizando cilindros.
Microrganismo: Staphylococcus epidermides ATCC 12228
Solução amostra: pesar, exatamente, cerca de 80 mg da amostra e transferir para balão volumétrico de 200 ml com auxílio de 120 ml deágua destilada. Agitar por 30 minutos e completar o volume comágua. Diluir, sucessivamente, até as concentrações de 4 μg/ml, 8μg/ml e 16 μg/ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), como diluente.
Solução padrão: pesar, exatamente, cerca de 20 mg de cloridrato de ciprofloxacino padrão, transferir para balão volumétrico de 50 ml e completar o volume com água. Diluir, sucessivamente, até as concentrações de 4 μg/ml, 8 μg/ml e 16 μg/ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) como diluente.
DOSEAMENTO
Proceder conforme descrito em Cromatografia líquida de alta eficiência (V.2.17.4) utilizando cromatógrafo provido de detector ultravioleta a 278 nm, coluna de 250 mm de comrimento e 4 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octadecilsilano, mantida a 30 °C; fluxo da fase móvel de 1,5 ml/minuto.
Fase móvel: mistura de ácido fosfórico 0,025 M com pH previamente
ajustado com trietilamina para 3,0±0,1 e acetonitrila (87:13).
Solução amostra: pesar, exatamente, cerca de 0,1 g de amostra, transferir
quantitativamente para balão volumétrico de 200 ml utilizando a
fase móvel como solvente. Agitar até solubilização, completar o volume
e homogeneizar.
Solução padrão: pesar, exatamente, cerca de 25 mg de cloridrato de ciprofloxacino padrão, transferir quantitativamente para balão volumétrico de 50 ml utilizando a fase móvel como solvente. Agitar até solubilização, completar o volume e homogeneizar.
Solução de resolução: dissolver na solução padrão quantidade da impureza ciprofloxacino etilenodiamina (cloridrato do ácido 1-ciclopropil- 6-fluor-1,4-diidro-4-oxo-7-[2-(aminoetil)amino]-3-quinolino carboxílico) para obter solução a 0,5 mg/ml.
A eficiência da coluna não deve ser menor do que 10 000 pratos teóricos/metro. Os tempos de retenção relativos são aproximadamente 0,7 para a impureza da solução de resolução e 1,0 para o cloridrato de ciprofloxacino. A resolução entre os picos do cloridrato de ciprofloxacino e da impureza não é inferior a 6. O desvio padrão relativo das áreas de replicatas dos picos registrados não deve ser maior que 1,5%.
Procedimento: injetar, separadamente, 10 μl das soluções amostra e padrão, registrar os cromatogramas e medir as áreas dos picos. Calcular o teor de C17H18FN3O3.HCl na solução amostra a partir das respostas obtidas para solução padrão e solução amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, à temperatura ambiente e protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antimicrobiano.
142
CLORIDRATO DE METOCLOPRAMIDA
Metoclopramidi hydrocloricum
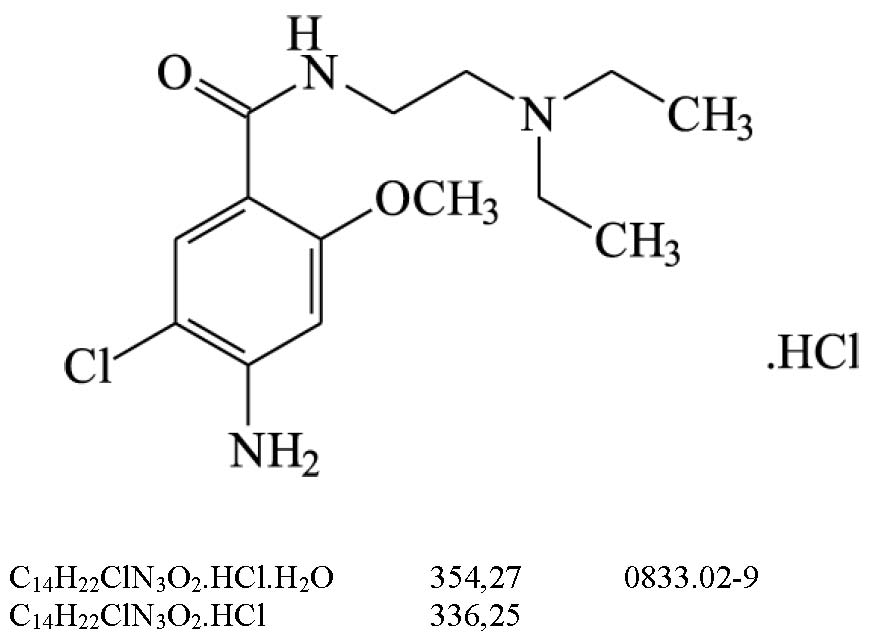
Monocloridrato de 4-amino-5-cloro-N-(2-dietilaminoetil)-2-metoxibenzamida
Contém, no mínimo, 99,0% e, no máximo, 101,0% de C14H22ClN3O2.HCl em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Cristais ou pó cristalino branco ou quase branco.
Solubilidade. Muito solúvel em água, solúvel em etanol, pouco solúvel em cloreto de metileno, praticamente insolúvel em éter.
IDENTIFICAÇÃO
O teste de identificação A pode ser omitido se forem realizados os testes B, C e D. Os testes de identificação B e C podem ser omitidos se forem realizados os testes A e D.
A. O espectro de absorção no infravermelho (V.2.14-4) da amostra dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de cloridrato de metoclopramida padrão, preparado de maneira idêntica.
B. Examinar, sob luz ultravioleta, o cromatograma obtido no ensaio de Substâncias relacionadas, antes de nebulizar com a solução de pdimetilaminobenzaldeído.A mancha principal obtida no cromatograma com a solução (2) corresponde em posição e intensidade àquela obtida com a solução (3).
C. Dissolver cerca de 2 mg da amostra em 2 ml de água. Prosseguir como descrito para a reação de identificação de Amina aromática primária (V.3.1.1).
D. Diluir 1 ml da solução obtida em Aspecto da solução para 2 ml com água. A solução responde às reações do íon cloreto (V.3.1.1).
ENSAIOS DE PUREZA
Aspecto da solução. Dissolver 2,5 g da amostra em água isenta de dióxido de carbono e diluir para 25 ml com o mesmo solvente. A solução é límpida e incolor.
pH (V.2.19). 4,5 e 6,0. Determinar na solução obtida em Aspecto da solução.
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando silica-gel GF254 como suporte, e mistura de hidróxido de amônio -dioxana-metanol-cloreto de metileno (2:10:14:90). Aplicar, separadamente, à placa, 5 μl de cada uma das soluções, recentemente preparadas, descritas a seguir.
Solução (1): dissolver 0,4 g da amostra em metanol e diluir para 10 ml com o mesmo solvente.
Solução (2): diluir l ml da solução (1) para 10 ml com metanol.
Solução (3): dissolver 20 mg de cloridrato de metoclopramida padrão em metanol e diluir para 5 ml com o mesmo solvente.
Solução (4): diluir 5 ml da solução (1) para 100 ml com metanol.
Diluir l ml desta solução para 10 ml com metanol.
Solução (5): dissolver 10 mg de N,N-dietiletilenodiamina em metanol e diluir para 50 ml com o mesmo solvente.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar.
Examinar sob luz ultravioleta (254 nm). Qualquer mancha no cromatograma obtido com a solução amostra (1), exceto a mancha principal não deve ser mais intensa do que a mancha obtida com a solução (4). Nebulizar com solução de p-dimetilaminobenzaldeído SR e deixar secar ao ar. Qualquer mancha no cromatograma obtido com a solução (1) que não tenha sido visualizada sob luz ultravioleta não é mais intensa do que a mancha obtida no cromatograma da solução (5).
Metais pesados (V.3.2.3). Utilizar 10 ml da solução obtida em Aspecto da solução. No máximo 0,002% (20 ppm).
Água (V.2.20.1). 4,5% a 5,5%. Determinar em 0,5 g de amostra.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,1%.
DOSEAMENTO
Dissolver 0,25 g da amostra em mistura de 5 ml de ácido clorídrico 0,01 M e 50 ml de etanol. Titular com hidróxido de sódio 0,1 M SV acompanhando a titulação potenciometricamente. Anotar o volume de hidróxido de sódio 0,1 M SV gasto entre os dois pontos de inflexão.
Cada ml de hidróxido de sódio 0,1 M SV equivale a 33,63 mg de C14H22ClN3O2.HCl.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, ao abrigo da luz.
ROTULAGEM
Observar a legislação vigente.
XII.2. REAGENTES E SOLUÇÕES REAGENTES
N,N-dietiletilenodiamina
Fórmula e massa molecular - C6H16N2 - 116,2
Descrição - Líquido de aparência levemente oleosa, incolor ou levemente amarelado, com forte odor amoniacal, irritante para a pele, olhos e membranas mucosas.
Características físicas - Densidade: 0,827. Faixa de ebulição 145 oC a 147 oC.
Água (V.2.20.1) - Determinar em 0,5 g. No máximo 1,0%.
p-Dimetilaminobenzaldeído SR
Preparação - Dissolver 0,2 g de p-dimetilaminobenzaldeído em 20 ml de etanol e adicionar 0,5 ml de ácido clorídrico. Agitar com carvão ativo e filtrar. Preparar imediatamente antes do uso.
143
CLORIDRATO DE PROPRANOLOL
Propranololi hydrochloridum
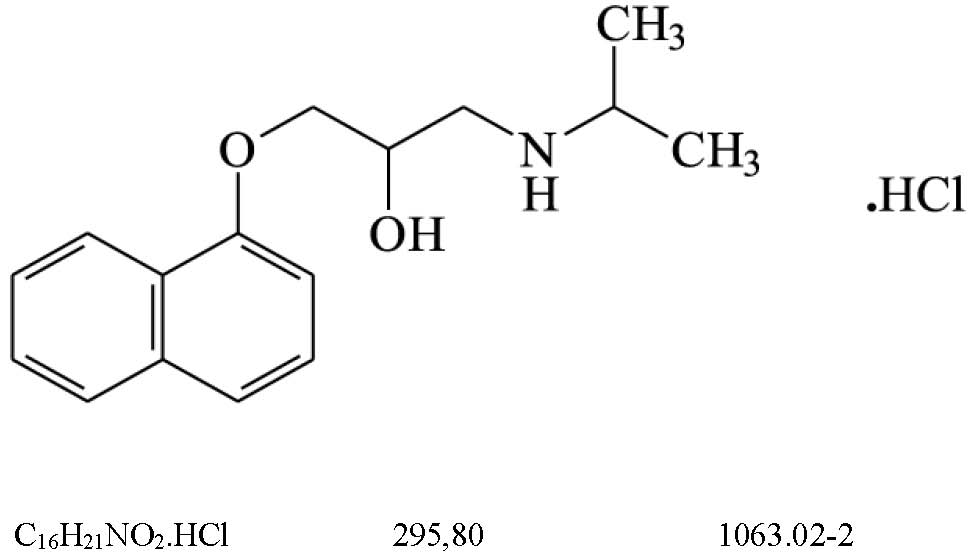
Cloridrato de (±)-1-isopropilamino-3-(1-naftiloxi)-2-propanol
Contém, no mínimo, 98,0% e, no máximo, 101,5% de C16H21NO2.HCl, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó branco ou quase branco, inodoro, sabor amargo e de aspecto cristalino ou amorfo.
Solubilidade. Solúvel em água e etanol, pouco solúvel em clorofórmio, insolúvel em éter etílico.
Constantes físico-químicas
Faixa de fusão (V.2.2): 163 °C a 166 °C.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4.) da amostra dessecada a 105 °C por 4 horas e dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de cloridrato de propranolol padrão, preparado de maneira idêntica.
B. Dissolver 0,1 g da amostra em 10 ml de água, transferir para funil de separação e alcalinizar com 3 ml de hidróxido de sódio M. Agitar, extrair com 10 ml de clorofórmio, agitando intensamente. Coletar a camada clorofórmica e lavar a camada aquosa com 3 ml de clorofórmio.
Reunir os extratos clorofórmicos e lavar com 5 ml de água.
Filtrar sobre sulfato de sódio anidro e evaporar até secura. Secar o resíduo sob vácuo a 60 ºC por 1 hora. O espectro de absorção no infravermelho (V.2.14.-4) do resíduo obtido, disperso em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro do resíduo, obtido após tratamento idêntico, realizado com 0,1 g de cloridrato de propranolol padrão.
C. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 200 nm a 400 nm, de uma solução a 0,004% (p/V) em metanol exibe máximos de absorção em 290, 306 e 319 nm, sendo as absorvâncias próximas de 0,84, 0,50 e 0,30, respectivamente.
D. Proceder conforme descrito em Substâncias relacionadas, utilizando mistura de metanol-amônia concentrada (99:1) como fase móvel e soluções a 1% (p/V) da amostra e do padrão, em metanol. A mancha principal obtida no cromatograma com a solução amostra corresponde em posição, cor e intensidade àquela obtida com a solução padrão.
E. O ponto de fusão do resíduo obtido no teste B de Identificação é cerca de 94 °C (V.2.2).
F. Responde às reações do íon cloreto (V.3.1.1).
ENSAIOS DE PUREZA
pH (V.2.19). 5,0 a 6,0. Determinar em solução a 1,0% (p/V).
Acidez e alcalinidade. Dissolver 0,2 g da amostra em água livre de dióxido de carbono e completar com água para 20 ml. Adicionar 0,2 ml de vermelho de metila SI e 0,2 ml de ácido clorídrico 0,01 M. A solução é vermelha. Adicionar 0,4 ml de hidróxido de sódio 0,01 M.
A solução é amarela.
Poder rotatório específico (V.2.8.). -1,0° a +1,0°. Determinar em solução aquosa a 4% (p/V) da substância dessecada.
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de tolueno-metanol (90:10) como fase móvel.
Aplicar, separadamente, à placa, 10 μl de cada uma das soluções metanólicas, recentemente preparadas, descritas a seguir.
Solução (1): solução a 10 % (p/V) da amostra.
Solução (2): solução a 0,02 % (p/V) da amostra.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar, observar sob luz ultravioleta (254 nm) e marcar as manchas. Nebulizar com solução contendo anisaldeído-acido acético glacial-metanol-ácido sulfúrico (0,5:10:85:5). Secar entre 100 °C e 105 °C.
Qualquer mancha secundária obtida no cromatograma com a solução (1) não é mais intensa que a mancha obtida com a solução (2).
Perda por dessecação (V.2.9). Determinar em estufa a 105 ºC, por 4 horas. No máximo 0,5%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g da amostra. No máximo 0,1% .
Metais pesados (V.3.2.3 - Método I). No máximo 0,002% (20 ppm).
DOSEAMENTO
A. Por titulação em meio não-aquoso (V.3.4.5). Dissolver 0,5 g da amostra em mistura de 50 ml de ácido acético glacial e 10 ml de acetato mercúrico a 6% (p/V) em ácido acético glacial. Titular comácido perclórico 0,1 M SV, determinando o ponto final potenciometricamente ou utilizando cloreto de metilrosanilínio SI (cristal violeta).
Realizar ensaio em branco e fazer as correções necessárias.
Cada ml do ácido perclórico 0,1 M equivale a 29,580 mg de C16H21NO2.HCl.
B. Por cromatografia líquida de alta eficiência. Proceder conforme descrito em Cromatografia líquida de alta eficiência (V.2.17.4), utilizando cromatógrafo provido de detector ultravioleta a 290 nm; coluna de 250 mm de comprimento e 4,6 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octilsilano (5 μm), mantida à temperatura ambiente; fluxo da fase móvel de 1,5 ml/minuto.
Fase móvel: dissolver 0,5 g de dodecil sulfato de sódio em 18 ml deácido fosfórico 0,15 M, adicionar 90 ml de acetonitrila, 90 ml de metanol e diluir com água para 250 ml.
Solução amostra: transferir, exatamente, cerca de 50 mg da amostra para balão volumétrico de 50 ml, adicionar 45 ml de metanol e levar ao ultra-som por 5 minutos. Completar com metanol, homogeneizar e filtrar. Transferir 5 ml desta solução para balão volumétrico de 25 ml, diluir com metanol, homogeneizar efiltrar em membrana de 0,45μm.
Solução padrão: transferir, exatamente, cerca de 50 mg de propranolol padrão para balão volumétrico de 50 ml com metanol, obtendo solução a 1 mg/ml. Transferir 5 ml para balão volumétrico de 25 ml, completar com metanol, homogeneizar e filtrar.
Solução de resolução: preparar solução a 0,25 mg/ml de cloridrato de procainamida em metanol. Transferir 5 ml desta solução e 5 ml da solução padrão para balão volumétrico de 25 ml. Diluir com metanol, homogeneizar e filtrar em membrana de 0,45 μm.
A resolução entre a procainamida e o cloridrato de propranolol não é menor que 2,0 e o fator de cauda do padrão não excede 3,0. O desvio padrão relativo das áreas de replicatas dos picos registrados não deve ser superior a 2%.
Procedimento: injetar, separadamente, 20 μl das soluções padrão e amostra, registrar os cromatogramas e medir as áreas dos picos.
Calcular o teor de C16H21NO2.HCl na solução amostra, a partir das respostas obtidas com as soluções padrão e amostra.
ARMAZENAMENTO
Em recipientes bem-fechados, protegido da luz.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Anti-hipertensivo, antiarrítmico, antianginoso.
143.1
CLORIDRATO DE PROPRANOLOL COMPRIMIDOS
Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C16H21NO2.HCl.
IDENTIFICAÇÃO
A. Pesar e pulverizar os comprimidos. Suspender em água quantidade do pó equivalente a 0,1 g de cloridrato de propranolol e filtrar.
Transferir o filtrado para funil de separação, alcalinizar com hidróxido de sódio 1 M e extrair com três volumes de 10 ml de éter etílico.
Lavar os extratos combinados com água até neutralizar. Filtrar sobre sulfato de sódio anidro, evaporar o filtrado e secar o resíduo sob vácuo a 50 ºC por uma hora. O espectro de absorção no infravermelho (V.2.14.-4) do resíduo, disperso em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles obtidos no espectro do resíduo obtido após tratamento idêntico realizado com 0,1 g do cloridrato de propranolol padrão.
B. O ponto de fusão do resíduo obtido no teste A de Identificação é de, aproximadamente, 94 ºC (V.2.2).
C. A solução a 0,004% (p/V) obtida no método A do Doseamento responde ao teste C de Identificação na monografia de cloridrato de propranolol.
D. Proceder conforme descrito em Substâncias relacionadas. A mancha principal obtida no cromatograma com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (3).
E. Suspender em água quantidade de pó dos comprimidos equivalente a 0,1 g de cloridrato de propranolol. Agitar e filtrar em papel de filtro adequado. O filtrado responde às reações do íon cloreto (V.3.1.1).
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). No máximo 30 minutos.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
Procedimento para uniformidade de conteúdo. Pesar, individualmente, e transferir cada comprimido para balão de 100 ml, adicionar 5 ml deácido clorídrico 1% (V/V), agitar até desintegração do comprimido.
Adicionar 70 ml de metanol e submeter ao ultra-som por 1 minuto.
Completar o volume com metanol, homogeneizar e filtrar em papel de filtro adequado, desprezando os primeiros mililitros. Diluir o filtrado até concentração de 0,004% (p/V). Preparar solução metanólica a 0,004% (p/V) de cloridrato de propranolol padrão e medir as absorvâncias das soluções resultantes em 290 nm (V.2.14.-3), utilizando metanol para ajuste do zero. Calcular o teor individual de C16H21NO2.HCl nos comprimidos a partir das leituras obtidas.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: ácido clorídrico 1% (V/V), 1 000 ml
Aparelhagem : cesta, 100 rpm
Tempo: 30 minutos
Procedimento: após o teste retirar alíquotas do meio de dissolução e diluir, se necessário, com ácido clorídrico 1% (V/V), até concentração adequada. Medir as absorvâncias em 289 nm (V.2.14.-3), utilizando o mesmo solvente para ajuste do zero. Calcular a quantidade de C16H21NO2.HCl dissolvido no meio, comparando as leituras obtidas com a da solução padrão na concentração de 0,004% (p/V), preparada no mesmo solvente.
Tolerância: não menos que 75% (T) da quantidade declarada de C16H21NO2.HCl se dissolvem em 30 minutos.
ENSAIOS DE PUREZA
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1) utilizando sílica-gel GF254, como suporte, e mistura de tolueno-metanol-amônia concentrada (80:20:01) como fase móvel. Aplicar, separadamente, à placa, respectivamente, 100, 20 e 100 μl de cada uma das soluções descritas a seguir.
Solução (1): solução a 1% (p/V) de cloridrato de propranolol padrão em metanol.
Solução (2): solução a 0,02% (p/V) cloridrato de propranolol padrão em metanol.
Solução (3): pesar e pulverizar os comprimidos. Pesar quantidade do pó equivalente a 50 mg de cloridrato de propranolol, transferir para balão volumétrico de 5 ml, completar com metanol, homogeneizar e filtrar, obtendo solução a 1% (p/V).
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar, observar sub luz ultravioleta (254 nm) e marcar as manchas. Nebulizar com solução contendo anisaldeído-acido acético glacial-metanol-ácido sulfúrico (0,5:10:85:5). Secar entre 100 °C e 105 °C.
Qualquer mancha secundária obtida no cromatograma com a solução (3) não é maior ou mais intensa que a mancha obtida com a solução (2).
DOSEAMENTO
A. Por espectrofotometria de absorção no ultravioleta. Pesar e pulverizar 20 comprimidos. Transferir quantidade do pó equivalente a 20 mg de cloridrato de propranolol, para balão volumétrico de 100 ml, adicionar 20 ml de água e agitar mecanicamente por 10 minutos.
Adicionar 50 ml de metanol e agitar por 10 minutos, completar o volume com metanol, homogeneizar e filtrar. Transferir, quantitativamente, 10 ml do filtrado para balão volumétrico de 50 ml, completando o volume com metanol. Preparar solução a 0,004% (p/V) de cloridrato de propranolol padrão em metanol e medir as absorvâncias das soluções em 290 nm (V.2.14.-3), utilizando metanol como branco.
Calcular a quantidade de C16H21NO2.HCl nos comprimidos a partir das leituras obtidas.
B. Por cromatografia líquida de alta eficiência. Prosseguir conforme descrito no item B de Doseamento na monografia de cloridrato de propranolol. Preparar a solução amostra como descrito a seguir.
Solução amostra: pesar e pulverizar não menos que 20 comprimidos.
Transferir exatamente, quantidade do pó equivalente a 50 mg de cloridrato de propranolol para balão volumétrico de 50 ml, adicionar 40 ml de metanol, agitar e deixar em banho de ultra-som por 5 minutos. Completar o volume com metanol, homogeneizar e filtrar.
Transferir 5 ml da solução anterior para balão volumétrico de 25 ml, completar o volume com metanol, homogeneizar e filtrar em membrana de 0,45 μm.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
144
DICLOFENACO SÓDICO
Diclofenacum natricum
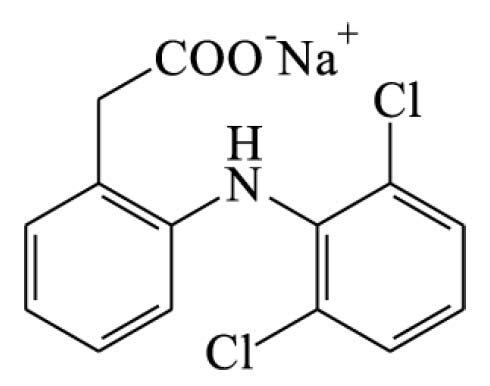
C14H10Cl2NNaO2 318,13 0398.03-9
2-[(2,6-Diclorofenil)amino]benzenoacetato de sódio
Contém, no mínimo, 99,0% e, no máximo, 101,0% de C14H10Cl2NNaO2, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó cristalino, branco a levemente amarelado, pouco higroscópico.
Solubilidade. Levemente solúvel em água, facilmente solúvel em metanol, solúvel em etanol, ligeiramente solúvel em ácido acético glacial, pouco solúvel em acetona, praticamente insolúvel em éter, clorofórmio e tolueno.
Constantes físico-químicas
Faixa de fusão (V.2.2): aproximadamente 280 ºC, com decomposição.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) da amostra dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro do diclofenaco sódico padrão, preparado de maneira idêntica.
B. Proceder conforme descrito no método B de Doseamento. O tempo de retenção do pico principal obtido com a solução amostra correspondeàquele do pico principal da solução padrão.
C. Dissolver 0,2 g da amostra em 50 ml de metanol e adicionar 1 ml de ácido nítrico. Produz-se coloração vermelha.
D. Dissolver 0,06 g da amostra em 0,5 ml de metanol e adicionar 0,5 ml de água. A solução responde às reações do íon sódio (V.3.1.1.- 5).
ENSAIOS DE PUREZA
Aspecto da solução. A solução utilizada no teste D em Identificação não é, significativamente, menos límpida do que um volume igual de metanol.
pH (V.2.19). 6,5 a 8,5. Determinar em solução aquosa a 1% (p/V).
Substâncias relacionadas. Proceder conforme descrito no método B de Doseamento.
Solução amostra: dissolver quantidade da amostra em diluente, de modo a obter solução contendo exatamente cerca de 0,75 mg/ml.
Solução padrão: dissolver quantidade do padrão de 1-(2,6-diclorofenil) indolin-2-ona em metanol, de modo a obter solução contendo exatamente cerca de 0,8 mg/ml. Diluir esta solução no diluente, de modo a obter solução contendo cerca de 4 μg/ml.
Solução de resolução: preparar como descrito no método B de Doseamento.
Procedimento: injetar, separadamente, 50 μl das soluções amostra e padrão, registrar os cromatogramas e medir as áreas dos picos. Calcular as porcentagens individuais das impurezas presentes, incluindo a 1-(2,6-diclorofenil)indolin-2-ona, pela expressão: (C/A)(Ra/Rp), onde:
C é a concentração em μg/ml de 1-(2,6-diclorofenil)indolin-2-ona;
A é quantidade em μg/ml de diclofenaco sódico encontrado na solução amostra utilizada no método B de doseamento; Ra é a resposta individual de cada impureza da solução amostra, incluindo a 1-(2,6- diclorofenil)indolin-2-ona, e Rp é área do pico do padrão de 1-(2,6- diclorofenil)indolin-2-ona. A porcentagem máxima tolerada é de 0,2% para cada impureza individual, incluindo a 1-(2,6-diclorofenil) indolin-2-ona e de, no máximo, 0,5% para a soma de todas as impurezas presentes.
Absorção de luz. Uma solução a 5% (p/V) da amostra em metanol é límpida a levemente amarelada e a absorvância em 440 nm é, no máximo, 0,05.
Metais pesados (V.3.2.3 - Método II). Pesar 2 g da amostra em um béquer de borossilicato de 100 ml. Incinerar a 500 ºC ou 600 ºC. Se o resíduo não se apresentar completamente branco após incineração, adicionar quantidade suficiente de peróxido de hidrogênio para solubilizar o resíduo e aquecer até completa evaporação. Repetir o procedimento até obter resíduo completamente branco. Proceder conforme descrito em Ensaio-limite para metais pesados iniciando a partir de “...esfriar, adicionar 4 ml de ácido clorídrico 6 M...”. No máximo 0,001% (10 ppm).
Perda por dessecação (V.2.9). Determinar em 1 g da amostra, em estufa entre 105 ºC e 110 ºC, por 3 horas. No máximo 0,5%.
DOSEAMENTO
A. Por titulação em meio não-aquoso (V.3.4.5). Dissolver 0,25 g da amostra em 30 ml de ácido acético glacial e titular com ácido perclórico 0,1 M SV, determinando o ponto final potenciometricamente.
Cada ml de ácido perclórico 0,1 M SV, equivale a 31,813 mg de C14H10Cl2NNaO2.
B. Por cromatografia líquida de alta eficiência (V.2.17.4), utilizando
cromatógrafo provido de detector ultravioleta a 254 nm; coluna de
150 mm e 4,6 mm de diâmetro interno, empacotada com sílica quimicamente
ligada a octilsilano (5 μm); fluxo da fase móvel de 1,0
ml/minuto.
Fase móvel: solução tampão fosfato pH 2,5-metanol (70:30);
Solução tampão fosfato pH 2,5: misturar iguais volumes de solução de ácido fosfórico 0,01 M e solução de fosfato de sódio monobásico 0,01 M. Se necessário, ajustar o pH para 2,5 ± 0,2.
Diluente: preparar mistura de metanol-água (70:30).
Solução amostra: dissolver quantidade exatamente pesada da amostra no diluente, de modo a obter solução contendo cerca de 0,75 mg/ml e diluir, quantitativamente, com o diluente para obter solução contendo 7,5 μg/ml.
Solução padrão: dissolver quantidade exatamente pesada do padrão de diclofenaco sódico em diluente, de modo a obter solução contendo cerca de 0,75 mg/ml e diluir, quantitativamente, com o diluente para obter solução contendo 7,5 μg/ml.
Solução de resolução: preparar uma solução no diluente, contendo 20μg/ml de ftalato de dietila, 7,5 μg/ml do padrão de 1-(2,6-diclorofenil) indolin-2-ona e 0,75 mg/ml do padrão de diclofenaco sódico.
A eficiência da coluna não é inferior a 16 000 pratos teóricos/metro;
os tempos de retenção relativos são de cerca de 0,5 para o ftalato de dietila, 0,6 para o 1-(2,6-diclorofenil)indolin-2-ona e 1 para o diclofenaco sódico. A resolução entre o ftalato de dietila e a 1-(2,6- diclorofenil)indolin-2-ona não é menor que 2,2 e entre a 1-(2,6- diclorofenil)indolin-2-ona e o diclofenaco sódico não é menor que 6,5. O desvio padrão relativo das áreas de replicatas dos picos registrados não é maior que 2,0%.
Procedimento: injetar, separadamente, 50 μl das soluções amostra e padrão, registrar os cromatogramas e medir as áreas dos picos. Calcular o teor de C14H10Cl2NNaO2 na solução amostra a partir das respostas obtidas para as soluções padrão e amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antiinflamatório.
144.1
DICLOFENACO SÓDICO COMPRIMIDOS
Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C14H10Cl2NNaO2. Devem apresentar revestimento gastro- resistente.
IDENTIFICAÇÃO
A. Remover o revestimento e pulverizar 10 comprimidos. Pesar o equivalente a 0,15 g de diclofenaco sódico, adicionar 0,5 ml de ácido acético e 15 ml de metanol. Deixar a suspensão em banho de ultrasom por 15 minutos e agitar durante 1 minuto. Filtrar e recolher o filtrado em 15 ml de água. Filtrar, o sólido formado sob pressão reduzida, lavá-lo com quatro porções de 5 ml de água e secá-lo a 105ºC por 2 a 3 horas. Solubilizar 50 mg de diclofenaco sódico padrão em 5 ml de metanol, adicionar 0,5 ml de ácido acético, 15 ml de água e agitar. Filtrar o sólido formado sob pressão reduzida, lavar com quatro porções de 5 ml de água e secar a 105 ºC por 2 a 3 horas. O espectro de absorção no infravermelho (V.2.14.-4) do resíduo obtido a partir dos comprimidos, disperso em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro do resíduo obtido a partir do diclofenaco sódico padrão.
B. Proceder conforme descrito no método B de Doseamento. O tempo de retenção do pico principal obtido com a solução amostra correspondeàquele do pico principal obtido com a solução padrão.
C. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 200 a 350 nm, de solução a 0,001% (p/V) em metanol, exibe máximos e mínimos somente nos mesmos comprimentos de onda de uma solução similar do diclofenaco sódico padrão.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste de desintegração para comprimidos com revestimento entérico.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
Procedimento para uniformidade de conteúdo. Proceder conforme descrito no método A de Doseamento.
TESTE DE DISSOLUÇÃO (V.1.5)
Etapa ácida
Meio de dissolução: ácido clorídrico 0,1 M, 900 ml
Aparelhagem: pás, 50 rpm
Tempo: 2 horas
Procedimento: após o teste, retirar os comprimidos ou a maior porção deles do interior da cuba, adicionar em cada uma, 20 ml de hidróxido de sódio 5 M e homogeneizar por 5 minutos. Em seguida, retirar alíquotas do meio de dissolução e filtrar. Medir as absorvâncias das soluções em 276 nm, utilizando solução contendo ácido clorídrico 0,1 M e hidróxido de sódio 5 M (9:2) para ajuste do zero. Calcular a quantidade de C14H10Cl2NNaO2 dissolvido no meio, comparando as leituras obtidas com a da solução padrão preparada como descrito a seguir. Pesar exatamente cerca de 68 mg de diclofenaco sódico padrão e transferir, quantitativamente, para balão volumétrico de 100 ml contendo 10 ml de hidróxido de sódio 0,1 M. Agitar até completa solubilização, completar o volume com água destilada e homogeneizar.
Transferir 2 ml desta solução para balão volumétrico de 100
ml, completar o volume com solução contendo ácido clorídrico 0,1 M
e hidróxido de sódio 5 M (9:2) e homogeneizar. Esta solução contém
0,00136 % (p/V) de diclofenaco sódico padrão.
Tolerância: não mais que 10% (T) da quantidade declarada de C14H10Cl2NNaO2 se dissolvem em 2 horas. Devem ser cumpridas as exigências especificadas na tabela de critérios de aceitação para a etapa ácida
Critérios de aceitação para etapa ácida
| Estágio | Número de unidades testadas | Critérios de aceitação |
|---|---|---|
| E1 | 6 | Nenhum valor individual é superior 10% do teor rotulado |
| E2 | 6 | A média das 12 unidades (E1 + E2) não é superior a 10% e nenhum valor individual é superior a 25% do teor rotulado |
| E3 | 12 | A média das 24 unidades (E1 + E2 + E3) não é superior a 10% e nenhum valor individual é superior a 25% do teor rotulado |
Etapa básica
Meio de dissolução: tampão fosfato pH 6,8, 900 ml
Aparelhagem: pás, 50 rpm
Tempo: 45 minutos
Tampão fosfato pH 6,8: dissolver 76 g de fosfato de sódio tribásico em água para obter 1 000 ml de solução. Misturar 250 ml desta solução com 750 ml de ácido clorídrico 0,1 M e, se necessário, ajustar o pH para 6,8 + 0,05 com ácido clorídrico 2 M ou hidróxido de sódio 2 M.
Procedimento: utilizar os mesmos comprimidos submetidos à etapaácida. Após o teste, retirar alíquota do meio de dissolução, filtrar e diluir em tampão fosfato pH 6,8 até concentração adequada. Medir as absorvâncias das soluções em 276 nm, utilizando o mesmo solvente para o ajuste do zero. Calcular a quantidade de C14H10Cl2NNaO2 dissolvido no meio, comparando as leituras obtidas com a da solução padrão preparada como descrito a seguir. Pesar exatamente cerca de 68 mg do diclofenaco sódico padrão e transferir, quantitativamente, para balão volumétrico de 100 ml contendo 10 ml de hidróxido de sódio 0,1 M. Agitar até completa solubilização, completar o volume com água destilada e homogeneizar. Transferir 3 ml desta solução para balão volumétrico de 100 ml, completar o volume com o meio de dissolução e homogeneizar. Esta solução contém 0,00204 % (p/V) de diclofenaco sódico padrão.
Tolerância: não menos que 75% (T) da quantidade declarada de C14H10Cl2NNaO2 se dissolvem em 45 minutos.
ENSAIOS DE PUREZA
Substâncias relacionadas. Por cromatografia líquida de alta eficiência (V.2.17.4). Utilizar as mesmas condições descritas no método B de Doseamento na monografia de diclofenaco sódico.
Solução amostra: pesar e pulverizar 20 comprimidos. Transferir quantidade do pó equivalente a 75 mg de diclofenaco sódico para balão volumétrico de 100 ml. Adicionar cerca de 50 ml de diluente. Deixar em banho de ultra-som por 15 minutos, completar o volume com o diluente, homogeneizar e filtrar.
Solução padrão: proceder conforme descrito em Substâncias relacionadas na monografia de diclofenaco sódico.
Solução de resolução: proceder conforme descrito em Substâncias relacionadas na monografia de diclofenaco sódico.
Procedimento: proceder conforme descrito em Substâncias relacionadas na monografia de diclofenaco sódico. A porcentagem máxima tolerada é de 1% para cada impureza individual, incluindo a 1-(2,6- diclorofenil)indolin-2-ona e de, no máximo, 1,5% para a soma de todas as impurezas presentes.
DOSEAMENTO
A. Por espectrofotometria no ultravioleta (V.2.14.-3). Pesar e pulverizar 20 comprimidos. Transferir quantidade do pó equivalente a 50 mg de diclofenaco sódico para balão volumétrico de 200 ml. Adicionar cerca de 100 ml de metanol. Deixar a solução em banho de ultra-som por 15 minutos, completar o volume com o mesmo solvente, homogeneizar e filtrar. Diluir, sucessivamente, em metanol, até concentração de 0,00125% (p/V). Preparar solução padrão na mesma concentração, utilizando o mesmo solvente. Medir as absorvâncias das soluções resultantes em 285 nm, utilizando metanol para ajuste do zero. Calcular o teor de C14H10Cl2NNaO2 na amostra, a partir das leituras obtidas.
B. Por cromatografia líquida de alta eficiência (V.2.17.4). Utilizar as mesmas condições descritas no método B de Doseamento na monografia de diclofenaco sódico.
Solução amostra: pesar e pulverizar 20 comprimidos. Transferir quantidade do pó equivalente a 75 mg de diclofenaco sódico para balão volumétrico de 100 ml. Adicionar cerca de 50 ml de diluente. Deixar em banho de ultra-som por 15 minutos, completar o volume com o diluente, homogeneizar e filtrar. Diluir, sucessivamente, com o diluente para obter solução contendo 7,5 μg/ml.
Solução padrão: dissolver quantidade exatamente pesada do padrão de diclofenaco sódico no diluente, de modo a obter solução contendo cerca de 0,75 mg/ml. Diluir, sucessivamente, com o diluente para obter solução contendo 7,5 μg/ml.
Solução de resolução: preparar conforme descrito no método B de Doseamento na monografia de diclofenaco sódico.
Procedimento: injetar, separadamente, 50 μl das soluções amostra e padrão, registrar os cromatogramas e medir as áreas dos picos. Calcular a quantidade de C14H10Cl2NNaO2 nos comprimidos, a partir das respostas obtidas com as soluções amostra e padrão.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
145
DIPIRONA
Dipironum
C13H16N3NaO4S.H2O 351,36 0439.01-0
C13H16N3NaO4S 333,34
Sal sódico monoidratado do ácido [(2,3-diidro-1,5-dimetil-3-oxo-2- fenil-1H-pirazol-4-il)metilamino]metanossulfônico
Contém, no mínimo, 98,0% e, no máximo, 101,0% de C13H16N3NaO4S, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó cristalino, quase branco e inodoro.
Solubilidade. Solúvel em água e em metanol, pouco solúvel em etanol, praticamente insolúvel em éter etílico, acetona, benzeno e clorofórmio.
IDENTIFICAÇÃO
A. A 2 ml de solução aquosa a 5% (p/V) adicionar 4 ml de ácido clorídrico e aquecer até a ebulição. Desprendem-se vapores sulfurosos.
Transferir 0,5 ml da solução aquecida para tubo de ensaio e,
em seguida, adicionar 0,5 ml de formaldeído e 1 ml do reagente de
Schiff. Desenvolve-se coloração violeta.
B. Misturar 50 mg com 1 ml de peróxido de hidrogênio 30% (p/p). A solução inicialmente desenvolve coloração azul, que desaparece rapidamente passando a vermelha intensa.
C. A 2 ml de solução aquosa 5% (p/V) adicionar 0,2 ml de ácido nítrico 6 M e 0,1 ml de nitrito de sódio 0,1% (p/V). Desenvolve-se coloração azul, que, em seguida, desaparece. Adicionar 0,5 ml de nitrato de prata 5% (p/V). Forma-se um precipitado branco, que se dissolve por agitação. A solução torna-se turva e se colore novamente de azul, passando lentamente para verde e depois para amarelo, com precipitação de prata metálica.
D. A 0,5 g da amostra adicionar 1 ml de ácido clorídrico 6 M.
Aquecer em chama oxidante. A chama adquire coloração amarela intensa.
ENSAIOS DE PUREZA
Aspecto e cor da solução. Dissolver 2,5 g da amostra em água isenta de dióxido de carbono e completar o volume para 50 ml com o mesmo solvente. A solução apresenta-se límpida. Imediatamente após a preparação, comparar 5 ml da solução com 5 ml da solução padrão, descrita a seguir. A cor não é mais intensa que a da solução padrão de cor.
Solução padrão de cor: misturar 0,75 ml da solução (1), 0,25 ml da solução (2), 0,25 ml da solução (3) e 48,75 ml da solução (4).
Solução (1): dissolver 4,51 g de cloreto férrico hexaidratado com 3,2 ml de ácido clorídrico 1 M e completar o volume com água para 100 ml.
Solução (2): dissolver 6,5 g de cloreto cobaltoso hexaidratado com 3 ml de ácido clorídrico 6 M e completar o volume com água para 100 ml.
Solução (3): dissolver 6,242 g de sulfato cúprico pentaidratado comágua e completar o volume para 100 ml.
Solução (4): ácido clorídrico 1% (p/V).
Acidez ou alcalinidade. Adicionar 0,1 ml de fenolftaleína SI a 5 ml da solução obtida em Aspecto e cor da solução. A cor da solução não sofre alteração. A viragem do indicador para rosa consome no máximo 0,1 ml de hidróxido de sódio 0,02 M.
Metais pesados (V.3.2.3 - Método II). No máximo 0,002% (20 ppm).
Sulfato (V.3.2.2). No máximo 0,1% (1 000 ppm).
Impurezas solúveis em clorofórmio. Pesar 1 g de amostra, adicionar 10 ml de clorofórmio, deixar em repouso durante 30 minutos. Filtrar e lavar duas vezes com 5 ml de clorofórmio. Evaporar em banhomaria e secar a 105 ºC até peso constante. No máximo 0,5%.
Perda por dessecação. Determinar em 0,25 g da amostra, em estufa a 105 ºC, até peso constante. No mínimo 4,9% e no máximo 5,3%.
DOSEAMENTO
Pesar, exatamente, cerca de 0,25 g da amostra e dissolver em 50 ml de água. Adicionar 3 ml de ácido acético 6% (V/V) e titular com solução de iodo 0,05 M SV em temperatura abaixo de 20 ºC, utilizando amido SI como indicador. Cada ml de iodo 0,05 M SV equivale a 16,67 mg de C13H16N3NaO4S ou a 17,57 mg de C13H16N3NaO4S.H2O.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Analgésico e antipirético.
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Preparação - Dissolver 1 g de fucsina básica em 600 ml de água gelada, adicionar 100 ml de sulfito monossódico 20% (p/V). Resfriar externamente Reagente de Schiff com gelo, sob agitação. Adicionar, lentamente, 10 ml de ácido clorídrico, diluir com água para 1 000 ml e filtrar. Se a solução escurecer, agitar com 0,2 a 0,3 g de carvão ativado até descoloração, filtrando imediatamente. Se ainda permanecer coloração rósea, adicionar 2 a 3 ml de ácido clorídrico e agitar.
Conservação - Deixar em repouso durante 12 horas antes da utilização, mantida ao abrigo da luz.
145.1
DIPIRONA COMPRIMIDOS
Contém, no mínimo, 95,0% e, no máximo, 105,0% da quantidade declarada de C13H16N3NaO4S.H2O.
IDENTIFICAÇÃO
A. Pesar e pulverizar os comprimidos. Adicionar a 0,5 g do pó, algumas gotas de peróxido de hidrogênio 30% (p/p). Desenvolve-se coloração azul, que desaparece rapidamente, passando a vermelha intensa.
B. Misturar 0,5 g do pó dos comprimidos com algumas gotas de persulfato de potássio 10% (p/V). Desenvolve-se coloração amarela intensa.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Desintegração (V.1.4.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: ácido clorídrico 0,1 M, 500 ml
Aparelhagem: pá, 50 rpm
Tempo: 45 minutos
Procedimento: após o teste, retirar alíquota do meio de dissolução, filtrar e diluir em ácido clorídrico 0,1 M até concentração adequada.
Medir as absorvâncias das soluções em 258 nm (V.2.14.-3), utilizando o mesmo solvente para ajuste do zero. Calcular a quantidade de C13H16N3NaO4S.H2O dissolvida no meio, comparando as leituras obtidas com a da solução padrão, na concentração de 0,001% (p/V), preparada no mesmo solvente.
Tolerância: não menos que 70% (T) da quantidade declarada de C13H16N3NaO4S.H2O se dissolvem em 45 minutos.
ENSAIOS DE PUREZA
Água (V.2.20.1). No máximo 3%.
DOSEAMENTO
Pesar e pulverizar 20 comprimidos. Pesar quantidade do pó equivalente a 0,25 g de C13H16N3NaO4S.H2O e transferir, quantitativamente, para erlenmeyer. Adicionar 25 ml de água, 5 ml de ácido acético glacial e agitar até dispersão homogênea. Titular com iodo 0,05 M SV, em temperatura abaixo de 15 ºC, utilizando 1 ml de amido SI, como indicador. Cada ml de iodo 0,05 M SV equivale a 17,57 mg de C13H16N3NaO4S.H2O.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
145.2
DIPIRONA SOLUÇÃO INJETÁVEL
Contém, no mínimo, 95,0% e, no máximo, 110,0% da quantidade declarada de C13H16N3NaO4S.H2O.
IDENTIFICAÇÃO
A. A 2 ml da solução injetável, adicionar 2 ml de peróxido de hidrogênio 30% (p/p). Desenvolve-se coloração azul, que desaparece rapidamente, passando a vermelha intensa.
B. A 2 ml da solução injetável, adicionar 2 ml de persulfato de potássio 10% (p/V). Desenvolve-se coloração amarela intensa.
C. Responde às reações do íon sódio.
CARACTERÍSTICAS
Determinação de volume (V.1.2). Cumpre o teste.
pH (V.2.19). 6,1 a 7,1.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1). Cumpre o teste.
DOSEAMENTO
Transferir volume da solução injetável correspondente a 5 g de C13H16N3NaO4S.H2O para balão volumétrico de 200 ml. Completar o volume com água e homogeneizar. Transferir 10 ml da solução para erlenmeyer, adicionar 50 ml de água, 5 ml de ácido acético glacial e homogeneizar. Titular com iodo 0,05 M SV, em temperatura abaixo de 15 ºC, utilizando amido SI como indicador. Cada ml de iodo 0,05 M SV equivale a 17,57 mg de C13H16N3NaO4S.H2O.
EMBALAGEM E ARMAZENAMENTO
Em recipientes de vidro tipo I, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
145.3
DIPIRONA SOLUÇÃO ORAL
Contém, no mínimo, 95,0% e, no máximo, 110,0% da quantidade declarada de C13H16N3NaO4S.H2O.
IDENTIFICAÇÃO
A solução oral responde aos testes A e B de Identificação na monografia de dipirona solução injetável.
CARACTERÍSTICAS
Determinação de volume (V.1.2). Cumpre o teste.
pH (V.2.19). 5,5 a 7,0.
TESTES DE SEGURANÇA BIOLÓGICA
Contagem de microrganismos viáveis totais (V.5.1.6.-1). Bactérias aeróbicas totais: no máximo 1 000 UFC/ml. Fungos e leveduras: no máximo 100 UFC/ml.
Pesquisa e identificação de patógenos (V.5.1.7). Cumpre o teste.
DOSEAMENTO
Transferir volume da solução oral correspondente a 5 g de
C13H16N3NaO4S.H2O para balão volumétrico de 200 ml. Completar o
volume com água e homogeneizar. Transferir 10 ml da solução para
erlenmeyer, adicionar 50 ml de água, 5 ml de ácido acético glacial e
homogeneizar. Titular com iodo 0,05 M SV, em temperatura abaixo
de 15 ºC, utilizando amido SI como indicador. Cada ml de iodo 0,05
M SV equivale a 17,57 mg de C13H16N3NaO4S.H2O.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
146
ETIONAMIDA
Ethionamidum
C8H10N2S 166,25 0506.01-X
2-Etil-4-piridinocarbotioamida
Contém, no mínimo, 98,0% e, no máximo, 102,0% de C8H10N2S, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó amarelo brilhante e com odor leve a moderado de sulfeto.
Solubilidade. Pouco solúvel em água, solúvel em metanol, ligeiramente
solúvel em etanol e em propilenoglicol e pouco solúvel em
clorofórmio e em éter etílico.
Constantes físico-químicas
Faixa de fusão (V.2.2): 158 oC a 164 oC.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) da amostra dessecada por 18 horas, sob sílica-gel e pressão reduzida e dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de etionamida padrão, preparada de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 220 nm a 350 nm, da solução amostra obtida no método B de Doseamento exibe máximos e mínimos somente nos mesmos comprimentos de onda da solução de etionamida padrão.
C. Dissolver 10 mg da amostra em 5 ml de metanol e adicionar 5 ml de nitrato de prata 0,1 M. Forma-se precipitado marrom escuro.
ENSAIOS DE PUREZA
pH (V.2.19). 6,0 a 7,0. Determinar em suspensão a 1% (p/V).
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de clorofórmio-metanol (90:10), como fase móvel. Aplicar, separadamente, à placa, 10 μl de cada uma das soluções, descritas a seguir, preparadas em acetona.
Solução (1): solução a 2% (p/V) da amostra.
Solução (2): solução a 0,01% (p/V) da amostra.
Solução (3): solução a 0,004% (p/V) da amostra.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar, examinar sob luz ultravioleta (254 nm). Nenhuma mancha secundária obtida no cromatograma com a solução (1) é mais intensa que aquela obtida com a solução (2) e não mais que uma mancha secudária obtida com a solução (1) é mais intensa que aquela obtida com a solução (3).
Perda por dessecação (V.2.9). Determinar em 1 g da amostra, em
estufa entre 100 oC e 105 oC, por 3 horas. No máximo 0,5%.
Cinzas sulfatadas (V.2.10). No máximo 0,2%.
DOSEAMENTO
A. Por titulação em meio não-aquoso (V.3.4.5). Pesar, exatamente, cerca de 0,25 g da amostra, dissolver em 50 ml de ácido acético glacial e adicionar 3 gotas de cloreto de metilrosanilínio SI (cristal violeta). Titular com ácido perclórico 0,1 M SV até viragem para laranja ou determinar o ponto final potenciometricamente. Realizar ensaio em branco e fazer as correções necessárias. Cada ml de ácido perclórico 0,1 M SV equivale a 16,625 mg de C8H10N2S.
B. Por espectrofotometria de absorção no ultravioleta. Dissolver 0,1 g da amostra em metanol e completar o volume para 100 ml com o mesmo solvente. Diluir, sucessivamente, em metanol, até concentração de 0,001% (p/V). Preparar solução padrão na mesma concentração, utilizando o mesmo solvente. Medir as absorvâncias das soluções resultantes em 290 nm (V.2.14.-3), utilizando metanol para ajuste do zero. Calcular o teor de C8H10N2S na amostra, a partir das leituras obtidas.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, em local fresco.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antibacteriano (tuberculostático).
146.1
ETIONAMIDA COMPRIMIDOS
Contém, no mínimo, 95,0% e, no máximo, 110,0% da quantidade declarada de C8H10N2S. Devem ser revestidos (revestimento açucarado).
IDENTIFICAÇÃO
A. Pesar e pulverizar os comprimidos. Agitar quantidade de pó equivalente a 0,125 g de etionamida com 25 ml de éter etílico, por 2 ou 3 minutos. Filtrar e deixar evaporar o filtrado à temperatura ambiente.
Secar o resíduo sob sílica-gel, utilizando pressão reduzida. O espectro de absorção no infravermelho (V.2.14.-4) do resíduo, disperso em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro da etionamida padrão, preparado de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 220 nm a 350 nm, da solução obtida no método B do Doseamento exibe máximo de absorção em 290 nm.
C. Pesar e pulverizar os comprimidos. Agitar quantidade de pó equivalente a 1 g de etionamida com 50 ml de metanol e filtrar utilizando papel de filtração lenta. Evaporar o filtrado em banho de vapor até secura. O resíduo obtido funde entre 155 ºC e 164 ºC.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Teste de desintegração (V.1.4.1). Utilizar ácido clorídrico 0,1 M. No máximo 30 minutos.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
ENSAIOS DE PUREZA
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de clorofórmio-metanol (9:1), como fase móvel. Aplicar, separadamente, à placa, 10 μl de cada uma das soluções descritas a seguir.
Solução (1): pesar e pulverizar os comprimidos. Agitar quantidade de pó, equivalente a 0,6 g de etionamida, com 20 ml de metanol. Adicionar metanol para completar 25 ml e filtrar.
Solução (2): diluir um volume da solução (1) para 200 volumes com metanol.
Desenvolver o cromatograma. Remover a placa, secar ao ar e observar sob luz ultravioleta (254 nm). Nenhuma mancha secundária obtida no cromatograma com a solução (1) é mais intensa que aquela obtida no cromatograma com a solução (2).
DOSEAMENTO
A. Por titulação em meio não-aquoso (V.3.4.5). Pesar e pulverizar 20 comprimidos. Agitar magneticamente, por 15 minutos, quantidade de pó, exatamente pesado, equivalente a cerca de 0,25 g de etionamida com 60 ml de ácido acético glacial. Adicionar 0,5 ml de anidrido acético e 4 gotas de cloreto de metilrosanilínio SI (cristal violeta).
Titular com ácido perclórico 0,1 M SV até viragem para laranja ou determinar o ponto final potenciometricamente. Realizar ensaio em branco e fazer as correções necessárias. Cada ml de ácido perclórico 0,1 M SV equivale a 16,625 mg de C8H10N2S.
B. Por espectrofotometria de absorção no ultravioleta. Pesar e pulverizar 20 comprimidos. Transferir para balão volumétrico de 250 ml, quantidade exatamente pesada, do pó, equivalente a cerca de 0,1 g de etionamida. Adicionar 200 ml de metanol, submeter ao banho de ultra-som por 10 minutos, agitar mecanicamente por 15 minutos e completar o volume com o mesmo solvente. Filtrar em papel de filtro, desprezando os primeiros 20 ml. Transferir 5 ml do filtrado para balão volumétrico de 200 ml, completar o volume com metanol e homogeneizar. Preparar solução de etionamida padrão na mesma concentração, utilizando o mesmo solvente. Medir as absorvâncias das soluções resultantes em 290 nm, utilizando metanol para ajuste do zero. Calcular a quantidade de C8H10N2S nos comprimidos a partir das leituras obtidas.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
147
EXTRATOS
Extracta
DEFINIÇÃO
Extratos são preparações de consistência líquida, sólida ou intermediária, obtidas a partir do material vegetal ou animal. O material utilizado na preparação de extratos pode sofrer tratamento preliminar, tal como inativação de enzimas, moagem ou desengorduramento.
Os extratos são preparados por percolação, maceração ou outro método adequado e validado, utilizando como solvente etanol, água ou outro solvente adequado. Após a extração, Materiais indesejáveis podem ser eliminados.
OBTENÇÃO
Percolação
Reduzir o material a ser extraído a partículas de tamanho adequado, misturar com parte do solvente preconizado para a extração e deixar em repouso por tempo não inferior a uma hora. Transferir o material umedecido para o percolador e percolar lentamente com o solvente preconizado, tomando o cuidado de verificar que o material no percolador esteja sempre coberto por solvente. A velocidade, o tempo e a temperatura de percolação, devem ser determinadas previamente para cada tipo de material vegetal. O resíduo de extração pode ser prensado e o líquido resultante da prensagem reunido ao percolado.
Maceração
Exceto quando prescrito diferentemente, reduzir o material a ser extraído a partículas de tamanho adequado, misturar com o solvente especificado e deixar em repouso, em recipiente fechado, pelo tempo determinado na monografia. Ao final do processo, o extrato é separado do resíduo e o resíduo prensado, reunindo-se o líquido resultante da prensagem com o extrato.
OBTENÇÃO DE EXTRATOS PADRONIZADOS
Extratos padronizados são extratos em que o teor de um ou mais constituintes é ajustado a valores previamente definidos. O ajuste do teor dos constituintes pode ser obtido por diluição do extrato com o solvente utilizado na extração ou com extratos mais diluídos, obtidos com o mesmo material e solvente, pela adição de materiais inertes ou por concentração. A concentração até a consistência e/ou teor de constituintes desejados é realizada por processos usuais, geralmente sob pressão reduzida, e em temperatura na qual a degradação dos constituintes do extrato é ausente ou mínima.
148
EXTRATOS FLUIDOS
Extracta fluida
DEFINIÇÃO
Extratos fluidos são preparações líquidas nas quais, exceto quando especificado diferentemente, uma parte do extrato, em massa ou volume, corresponde a uma parte, em massa, da droga seca, utilizada na sua preparação. Se necessário, os extratos fluidos podem ser padronizados, em termos de concentração do solvente, teor de constituintes ou resíduo seco. Se necessário, podem ser adicionados de conservantes inibidores do crescimento microbiano.
OBTENÇÃO
Os extratos fluidos podem ser obtidos por percolação, maceração ou por dissolução de extratos secos ou moles utilizando como solvente unicamente etanol, água ou misturas etanol/água de proporção adequada.
Se necessário, o extrato obtido pode ser filtrado. Qualquer que seja o processo de obtenção, os extratos fluidos apresentam composição e características comparáveis. A formação de um ligeiro sedimento durante a armazenagem é aceitável, desde que a composição do extrato não sofra modificações significativas.
ENSAIOS DE PUREZA
Densidade relativa (V.3.3.1). Quando for o caso, os extratos fluidos devem cumprir com os limites prescritos na monografia.
Determinação de álcool (V.3.4.8). Determinar teor de álcool em extratos fluidos obtidos com etanol ou misturas etanol/água. O teor deálcool deve cumprir o especificado na monografia.
Determinação de metanol e 2-propanol (V.4.3.1). A não ser quando especificado diferentemente, os extratos fluidos devem conter não mais de 0,05% (V/V) de metanol e não mais de 0,05% (V/V) de 2- propanol.
Resíduo seco. Transferir 2 ml ou 2 g de extrato fluido para pesafiltros
ou placa de Petri, medindo, aproximadamente, 50 mm em
diâmetro e 30 mm de altura. Evaporar até secura em banho-maria e
dessecar em estufa a 100 oC-105 oC por 3 horas. Deixar esfriar em
dessecador, sobre pentóxido de fósforo e pesar. Calcular o resíduo
seco em porcentagem sobre a massa ou sobre o volume.
EMBALAGEM E ARMAZENAMENTO
Armazenar em recipientes bem-fechados, ao abrigo da luz.
ROTULAGEM
O rótulo deve conter as seguintes informações:
o nome da droga que deu origem ao extrato;
se o extrato foi preparado com droga fresca (quando for o caso);
composição do solvente e o teor de etanol em porcentagem (V/V) no solvente utilizado na preparação;
quando for o caso o teor de etanol em porcentagem (V/V) no produto final;
teor de princípios ativos e/ou relação droga/extrato final;
nome e concentração de conservantes antimicrobianos adicionados.
149
EXTRATOS MOLES
Extracta spissa
DEFINIÇÃO
Os extratos moles são preparações de consistência pastosa obtidos por evaporação parcial do solvente utilizado na sua preparação. São obtidos utilizando-se como solvente unicamente etanol, água ou misturas etanol/água de proporção adequada. Apresentam no mínimo 70% de resíduo seco (p/p). Os extratos moles podem ser adicionados de conservantes para inibir crescimento microbiano.
ENSAIOS DE PUREZA
Resíduo seco. Pesar rapidamente em pesa-filtro ou em placa de Petri medindo aproximadamente 50 mm em diâmetro e 30 mm de altura, 2 g de extrato mole. Evaporar até secura em banho-maria e dessecar em estufa a 100 oC-105 oC por 3 horas. Deixar esfriar no dessecador, sobre pentóxido de fósforo e pesar. Calcular o resíduo seco em porcentagem sobre a massa. Alternativamente, determinar o resíduo seco em 2 g de extrato mole em balança de determinação de umidade.
EMBALAGEM E ARMAZENAMENTO
Armazenar em recipientes bem-fechados, ao abrigo da luz.
ROTULAGEM
O rótulo deve conter as seguintes informações:
o nome da droga que deu origem ao extrato;
nome e quantidade do material inerte utilizado;
se o extrato foi preparado com droga fresca (quando for o caso);
composição do solvente e o teor de etanol em porcentagem (V/V) no extrato líquido que lhe deu origem;
teor de princípios ativos e/ou relação droga/extrato final;
nome e concentração de conservantes antimicrobianos adicionados.
150
EXTRATOS SECOS
Extracta sicca
DEFINIÇÃO
Extratos secos são preparações sólidas obtidas pela evaporação do solvente utilizado na sua preparação. Apresentam, no mínimo, 95% de resíduo seco, calculados como percentagem de massa. Os extratos secos podem ser adicionados de materiais inertes adequados. Os extratos secos padronizados têm o teor de seus constituintes ajustado pela adição de materiais inertes adequados ou pela adição de extratos secos obtidos com a mesma droga utilizada na preparação. Quando necessário, a monografia poderá prescrever realização de ensaio limite para o solvente utilizado na preparação.
ENSAIOS DE PUREZA
Resíduo seco. Pesar rapidamente, numa placa de Petri medindo, aproximadamente, 50 mm em diâmetro e 30 mm de altura, 0,50 g de extrato seco finamente pulverizado. Dessecar em estufa a 100 oC-105 oC por 3 horas. Deixar esfriar no dessecador, sobre pentóxido de fósforo e pesar. Calcular o resíduo seco em porcentagem sobre a massa.
EMBALAGEM E ARMAZENAMENTO
Armazenar em recipientes hermeticamente fechados, ao abrigo da luz.
ROTULAGEM
O rótulo deve conter:
o nome da droga que deu origem ao extrato;
nome e quantidade do material inerte utilizado;
se o extrato foi preparado com droga fresca (quando for o caso);
nome do solvente e o teor de etanol em percentagem (V/V) no solvente utilizado na preparação;
teor de princípios ativos e/ou relação droga/extrato final.
151
FLUORETO DE SÓDIO
Natrii fluoridum
NaF 41,99 1113.23- 2
Contém, no mínimo, 98,0% e, no máximo, 102,0% de NaF, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó branco e inodoro.
Solubilidade. Solúvel em água, insolúvel em etanol.
IDENTIFICAÇÃO
A. Transferir 1 g para cadinho de platina, em capela, e adicionar 15 ml de ácido sulfúrico. Cobrir com uma peça de vidro límpida e polida e aquecer em banho-maria por 1 hora. Retirar a tampa de vidro, lavar com água e secar. A superfície do vidro fica marcada.
B. A solução a 4% (p/V) responde às reações para o íon sódio
( V. 3.1.1.- 5).
ENSAIOS DE PUREZA
Acidez ou alcalinidade. Dissolver 2 g da amostra em 40 ml de água numa cápsula de platina, adicionar 10 ml de uma solução saturada de nitrato de potássio, resfriar a solução a 0 ºC e adicionar 3 gotas de fenolftaleína SI. Se a solução adquirir coloração rosa, deve tornar-se incolor pela adição de, no máximo, 0,5 ml de ácido sulfúrico 0,05 M.
Se nenhuma coloração aparecer, adicionar, no máximo, 2 ml de hidróxido de sódio 0,1 M. Desenvolve-se coloração rosa que persiste por 15 segundos.
Fluorossilicato. Aquecer a solução neutralizada, obtida no ensaio de Acidez e alcalinidade e titular, ainda quente, com hidróxido de sódio 0,1 M SV até coloração rosa permanente. São necessários, no máximo, 1,5 ml de hidróxido de sódio 0,1 M SV.
Cloretos. Dissolver 0,3 g da amostra em 20 ml de água e adicionar 0,2 g de ácido bórico, 1 ml de ácido nítrico SR e 1 ml de nitrato de prata 0,1 M. Qualquer turvação resultante não é mais intensa do que a de uma preparação contendo 0,2 ml de ácido clorídrico 0,01 M, obtida nas mesmas condições. No máximo 0,012% (120 ppm). Metais pesados (V.3.2.3 - Método I). Transferir 1 g da amostra para cápsula ou cadinho de platina, adicionar 1 ml de água e 3 ml de ácido sulfúrico, aquecer, em capela, a temperatura tão baixa quanto possível, até que todo ácido sulfúrico tenha sido expelido. Dissolver o resíduo em 20 ml de água, neutralizar a solução com hidróxido de amônio usando fenolftaleína SI. Adicionar 1 ml de ácido acético glacial, diluir com água para 35 ml e filtrar. Usar o filtrado para o ensaio. No máximo 0,002% (20 ppm).
Perda por dessecação (V.2.9). Determinar em 1 g da amostra, em estufa a 150 ºC, por 4 horas. No máximo 1%.
DOSEAMENTO
Proceder conforme descrito em titulação em meio não-aquoso (V.3.4.5). Pesar, exatamente, cerca de 80 mg da amostra, adicionar 5 ml de anidrido acético e 20 ml de ácido acético glacial e aquecer brandamente até completa dissolução. Deixar esfriar e adicionar 20 ml de dioxano. Adicionar 3 gotas de cloreto de metilrosanilínio SI (cristal violeta) e titular com ácido perclórico 0,1 M SV até coloração verde esmeralda. Realizar ensaio em branco e fazer as correções necessárias. Cada ml de ácido perclórico 0,1 M equivale a 4,199 mg de NaF.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Profilático dental.
152
FUROSEMIDA
Furosemidum
Ácido 5-(aminossulfonil)-4-cloro-2-[(2-furanilmetil)amino]benzóico
Contém, no mínimo, 98,0% e, no máximo, 101,0% de C12H11 ClN2O5S, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó cristalino branco ou levemente amarelo, inodoro.
Solubilidade. Praticamente insolúvel em água, facilmente solúvel em acetona e dimetilformamida, solúvel em metanol, pouco solúvel em etanol e éter etílico, praticamente insolúvel em clorofórmio. Solúvel em soluções aquosas de hidróxidos alcalinos.
Constantes físico-químicas
Ponto de fusão (V.2.2): em torno de 210 ºC, com decomposição.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14-4) da amostra dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de furosemida padrão, preparado de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14-3) de uma solução a 0,0005% (p/V), em hidróxido de sódio 0,1 M, exibe máximos em 228, 271 e 333 nm, idênticos aos observados no espectro de solução similar de furosemida padrão. As absorvâncias das soluções padrão e amostra em 271 nm não diferem mais que 3%, quando calculadas em relação à substância dessecada.
C. Dissolver cerca de 5 mg em 10 ml de metanol. Transferir 1 ml desta solução para balão de refluxo, adicionar 10 ml de ácido clorídrico 2 M e submeter a refluxo por 15 minutos. Esfriar e adicionar 15 ml de hidróxido de sódio 1 M e 5 ml de nitrito de sódio 0,1% (p/V). Homogeneizar e aguardar por 3 minutos. Adicionar 5 ml de sulfamato de amônio 2,5% (p/V), homogeneizar e adicionar 1 ml de solução recém-preparada de dicloridrato de N-(1-naftil)etilenodiamina 0,5% (p/V). Desenvolve-se coloração vermelho-violeta.
ENSAIOS DE PUREZA
Aminas primárias aromáticas livres. Transferir 0,1 g da amostra para balão volumétrico de 25 ml, com auxílio de metanol. Agitar, completar o volume com metanol, homogeneizar e filtrar. Pipetar 1 ml do filtrad, transferir para balão volumétrico de 25 ml, adicionar, com agitação, 3 ml de dimetilformamida, 12 ml de água destilada e 1 ml de ácido clorídrico M. Esfriar e adicionar 1 ml de nitrito de sódio 0,5% (p/V), com agitação. Deixar em repouso durante 5 minutos.
Adicionar 1 ml de ácido sulfâmico 2,5% (p/V) com agitação e deixar em repouso por 3 minutos. Em seguida, adicionar 1 ml de solução de dicloridrato de N-(1-naftil)etilenodiamina 0,5% (p/V) e diluir para 25 ml com água destilada. Realizar ensaio em branco, em paralelo, nas mesmas condições, substituindo 1 ml do filtrado por 1 ml de metanol.
Realizar imediatamente a leitura da absorvância, em 530 nm. A absorvância obtida não é superior a 0,20.
Cloretos (V.3.2.1). A 1 g da amostra, adicionar mistura de 0,2 ml deácido nítrico e 30 ml de água. Agitar durante 5 minutos, deixar em repouso durante 15 minutos e filtrar. Utilizar 15 ml do filtrado. No máximo 0,02% (200 ppm).
Sulfatos (V.3.2.2). A 2 g da amostra, acrescentar mistura de 0,2 ml deácido acético e 30 ml de água. Agitar durante 5 minutos, deixar em repouso por 15 minutos e filtrar. Utilizar 15 ml do filtrado. No máximo 0,03% (300 ppm).
Metais pesados (V.3.2.3 - Método II). Utilizar 1 g de amostra. No máximo 0,002% (20 ppm).
Perda por dessecação (V.2.9). Determinar em estufa a 105 oC, por 3 horas. No máximo 1%.
Cinzas sulfatadas (V.2.10). No máximo 0,1%.
DOSEAMENTO
Dissolver 0,25 g da amostra em 20 ml de dimetilformamida, adicionar 0,2 ml de solução de azul de bromotimol 1% (p/V) em dimetilformamida e titular com hidróxido de sódio 0,1 M SV até coloração azul. Realizar ensaio em branco e fazer as correções necessárias.
Cada ml de hidróxido de sódio 0,1 M SV equivale a 33,07 mg de C12H11 ClN2O5S.
EMBALAGEM E ARMAZENAMENTO
Em recipientes opacos bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Diurético.
152.1
FUROSEMIDA COMPRIMIDOS
Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C12H11 ClN2O5S.
IDENTIFICAÇÃO
O espectro de absorção no ultravioleta (V.2.14..-3), na faixa de 220 nm a 340 nm, da solução final obtida no Doseamento exibe máximos de absorção em 228 nm e 271 nm.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
Procedimento para uniformidade de conteúdo. Pesar, separadamente, cada comprimido, e triturar. Transferir, quantitativamente, para balão volumétrico de 100 ml, adicionar hidróxido de sódio 0,1 M, agitar, completar o volume com o mesmo solvente e homogeneizar. Filtrar, transferir 1 ml do filtrado para balão volumétrico de 50 ml e completar o volume com o mesmo solvente. Preparar solução padrão na mesma concentração, utilizando o mesmo solvente. Medir as absorvâncias em 271 nm (V.2.14.-3), utilizando hidróxido de sódio 0,1 M para ajuste do zero. Calcular a quantidade de C12H11 ClN2O5S no comprimido, a partir das leituras obtidas.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: tampão fosfato pH 5,8, 900 ml
Aparelhagem: pás, 50 rpm
Tempo: 30 minutos
Procedimento: após o teste, retirar alíquota do meio de dissolução, filtrar e diluir com tampão fosfato pH 5,8 até concentração adequada. Medir as absorvâncias das soluções em 271 nm (V.2.14-3), utilizando o mesmo solvente, para ajuste do zero. Calcular a quantidade de C12H11 ClN2O5S dissolvida no meio, comparando as leituras obtidas com a da solução padrão na concentração de 0,0008% (p/V) preparada no mesmo solvente.
Tolerância: não menos que 80% (T) da quantidade declarada de C12H11 ClN2O5S se dissolvem em 30 minutos.
ENSAIOS DE PUREZA
Aminas aromáticas primárias livres. Pulverizar os comprimidos e pesar do pó o equivalente a 0,1 g de furosemida. Transferir para balão volumétrico de 25 ml, com auxílio de metanol. Agitar, completar o volume com metanol, homogeneizar e filtrar. Prosseguir como descrito em Aminas aromáticas primárias livres, na monografia de furosemida, a partir de “Pipetar 1 ml do filtrado...”. A absorvância obtida não é superior a 0,20.
DOSEAMENTO
Pesar e pulverizar 20 comprimidos. Transferir quantidade de pó equivalente a 0,2 g de furosemida para balão volumétrico de 500 ml com auxílio de 300 ml de hidróxido de sódio 0,1 M. Agitar por 10 minutos. Completar o volume com o mesmo solvente, homogeneizar e filtrar. Diluir 5 ml do filtrado para 250 ml com hidróxido de sódio 0,1 M e homogeneizar. Preparar solução padrão na mesma concentração, utilizando o mesmo solvente. Medir as absorvâncias das soluções resultantes em 271 nm (V.2.14.-3), utilizando hidróxido de sódio 0,1 M para ajuste do zero. Calcular o conteúdo de C12H11 ClN2O5S nos comprimidos a partir das leituras obtidas. Alternativamente realizar o cálculo utilizando A(1%,1cm) = 580, em 271 nm.
EMBALAGEM E ARMAZENAMENTO
Em recipientes opacos bem-fechados.
ROTULAGEM
Observar a legislação vigente.
152.2
FUROSEMIDA SOLUÇÃO INJETÁVEL
Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C12H11 ClN2O5S.
IDENTIFICAÇÃO
Transferir, para balão volumétrico de 100 ml, volume da solução injetável equivalente a 20 mg de furosemida, completar o volume com água e homogeneizar. Diluir 5 ml da solução para 100 ml com hidróxido de sódio 0,1 M. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 220 nm a 340 nm, da solução obtida, exibe máximos em 228 nm e 271 nm.
CARACTERÍSTICAS
Determinação de volume (V.1.2). Cumpre o teste.
pH (V.2.19). 8,0 a 9,3.
ENSAIOS DE PUREZA
Aminas primárias aromáticas livres. Pipetar volume da solução injetável equivalente a 0,1 g de furosemida, transferir para balão volumétrico de 25 ml, completar o volume com metanol, homogeneizar e filtrar. Prosseguir como descrito em Aminas aromáticas primárias livres, na monografia de furosemida, a partir de “Pipetar 1 ml do filtrado...”. A absorvância obtida não é superior a 0,20.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1). Cumpre o teste.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 1 ml/kg, empregando solução de furosemida a 0,2 mg/ml em solução fisiológica.
DOSEAMENTO
Pipetar volume da solução injetável equivalente a 20 mg de furosemida, transferir para balão volumétrico de 100 ml, completar o volume com água e homogeneizar. Diluir 5 ml do filtrado para 100 ml com hidróxido de sódio 0,1 M e homogeneizar. Preparar solução padrão na mesma concentração em hidróxido de sódio 0,1 M. Medir as absorvâncias das soluções resultantes em 271 nm (V.2.14.-3), utilizandohidróxido de sódio 0,1 M para ajuste do zero. Calcular o conteúdo de C12H11 ClN2O5S na solução injetável a partir das leituras obtidas. Alternativamente, realizar o cálculo utilizando A(1%,1cm) = 580, em 271 nm.
EMBALAGEM E ARMAZENAMENTO
Em recipientes de vidro tipo I, protegidos da luz.
153
GLIBENCLAMIDA
Glibenclamidum
5-Cloro-N-[2-[4-[[[(cicloexilamino)carbonil]amino]sulfonil]fenil]etil]- 2-metoxibenzamido
Contém, no mínimo, 98,0% e, no máximo, 102,0% de C23H28ClN3O5S, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó cristalino, branco ou quase branco, inodoro ou quase inodoro.
Solubilidade. Praticamente insolúvel em água, solúvel em dimetilformamida, ligeiramente solúvel em diclorometano, pouco solúvel em etanol, metanol e clorofórmio, praticamente insolúvel em éter etílico.
Constantes físico-químicas
Faixa de fusão (V.2.2): 169 °C a 174 °C.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) da amostra previamente dessecada e dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de glibenclamida padrão, preparado de maneira idêntica.
B. Dissolver cerca de 50 mg da amostra em metanol, submeter ao banho de ultra-som, se necessário, e completar o volume com metanol para 50 ml. Transferir 10 ml da solução para balão volumétrico de 100 ml, adicionar 1 ml de ácido clorídrico M e completar o volume com metanol. O espectro de absorção no ultravioleta (V.2.14.- 3) na faixa de 230 nm a 350 nm, da solução final, exibe máximos em 300 nm e em 275 nm. A absorvância a 300 nm é de 0,61 a 0,65 e a 275 nm é de 0,27 a 0,32.
C. Proceder conforme descrito em Substâncias relacionadas. A mancha principal obtida no cromatograma com a solução (l) corresponde em posição, tamanho e coloração à mancha principal obtida no cromatograma com a solução (3).
D. Ferver 50 mg da amostra com 1 ml de hidróxido de sódio 6 M. Os vapores que se desprendem após evaporação da água mudam, de vermelho para azul, o papel tornassol umedecido e apresentam odor irritante característico das aminas.
E. Misturar 0,2 g da amostra com 0,25 g de carbonato de sódio anidro e 0,25 g de carbonato de potássio. Incinerar a mistura por 10 minutos, esfriar, adicionar ao resíduo l0 ml de água quente, agitar por um minuto e filtrar. O filtrado responde às reações dos íons cloreto e sulfato (V.3.1.1).
ENSAIOS DE PUREZA
Cor da solução (V.2.12). Uma solução a l% (p/V) em etanol, preparada com aquecimento, é límpida e incolor.
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de clorofórmio-cicloexano-etanol-ácido acético glacial (45:45:5:5), como fase móvel. Aplicar, separadamente, à placa, l0 μl de cada uma das soluções descritas a seguir.
Solução (1): solução a 2% (p/V) da amostra em mistura de clorofórmio- metanol (1:1).
Solução (2): solução a 0,004% (p/V) da amostra em mistura de clorofórmio-metanol (1:1).
Solução (3): dissolver 0,2 g de glibenclamida padrão em l0 ml de metanol e aquecer sob refluxo por l0 minutos.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e examinar sob luz ultravioleta (254 nm). Nenhuma mancha secundária obtida no cromatograma com a solução (1) é mais intensa que a mancha obtida com a solução (2). O teste só é válido se o cromatograma obtido com a solução (3) apresentar duas manchas nitidamente separadas.
Metais pesados (V.3.2.3 - Método II). No máximo 0,002% (20 ppm).
Perda por dessecação (V.2.9). Determinar em l g da amostra, em estufa a 105 oC, por 6 horas. No máximo 1%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo0,5%.
DOSEAMENTO
Pesar, exatamente, cerca de 0,5 g de amostra e dissolver em 100 ml
de etanol, previamente neutralizado com hidróxido de sódio 0,1 M
SV, utilizando fenolftaleína SI como indicador. Titular com hidróxido
de sódio 0,1 M SV. Cada ml de hidróxido de sódio 0,1 M SV
equivale a 49,401 mg de C23H28ClN3O5S.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz e em local fresco.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Hipoglicemiante oral.
ROTULAGEM
Observar a legislação vigente.
153.1
GLIBENCLAMIDA COMPRIMIDOS
Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C23H28ClN3O5S.
IDENTIFICAÇÃO
A. Pesar e pulverizar os comprimidos. Misturar em gral quantidade de pó equivalente a cerca de 20 mg de glibenclamida com 20 ml de mistura de cloreto de metileno-acetona (20:10). Filtrar e evaporar o filtrado à temperatura ambiente. Secar o resíduo obtido a 105 °C por duas horas. O espectro de absorção no infravermelho (V.2.14.-4) do resíduo, disperso em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro da glibenclamida padrão.
B. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 230 a 350 nm, da solução obtida no Doseamento exibe máximos em 275 nm e 300 nm .
C. Proceder conforme descrito em Substâncias relacionadas. A mancha principal obtida no cromatograma com a solução (1) corresponde em posição, tamanho e coloração à mancha principal obtida no cromatograma com a solução (3).
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). No máximo 15 minutos.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
Procedimento para uniformidade de conteúdo. Pesar individualmente e transferir cada comprimido para balão volumétrico de 50 ml. Adicionar 1 ml de ácido clorídrico 0,5 M e agitar até o comprimido se desintegrar. Adicionar 30 ml de metanol, submeter ao banho de ultrasom por 15 minutos e, em seguida, agitar mecanicamente por mais l5 minutos. Completar o volume com metanol e homogeneizar. Filtrar.
Preparar a solução padrão e o branco conforme descrito no Doseamento.
Medir as absorvâncias das soluções resultantes em 300 nm (V.2.14.-3), utilizando o branco para ajuste do zero. Calcular o teor de C23H28ClN3O5S em cada comprimido a partir das leituras obtidas.
ENSAIOS DE PUREZA
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.-1), utilizando sílica-gel GF254, como suporte, e mistura de clorofórmio-cicloexano-etanol-ácido acético glacial (45:45:5:5), como fase móvel. Aplicar separadamente à placa 10 μl de cada uma das soluções descritas a seguir.
Solução (1): misturar em gral quantidade de pó dos comprimidos
equivalente a 40 mg de glibenclamida com 20 ml de mistura de
cloreto de metileno-acetona (20:10) e filtrar. Evaporar o filtrado até
secura à temperatura não excedente a 40 °C e sob pressão reduzida.
Dissolver o resíduo em 4 ml de clorofórmio-metanol (1:1).
Solução (2): solução a 0,024% (p/V) de glibenclamida padrão em clorofórmio-metanol (1:1).
Solução (3): solução a 1,% (p/V) de glibenclamida padrão em clorofórmio- metanol (1:1).
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e examinar sob luz ultravioleta (254 nm). Nenhuma mancha secundária no cromatograma com a solução (1) é mais intensa que a mancha obtida com a solução (2).
DOSEAMENTO
Pesar e pulverizar 20 comprimidos. Transferir, quantitativamente, para balão volumétrico de 200 ml, quantidade do pó, exatamente pesada, equivalente a cerca de 20 mg de glibenclamida. Adicionar 4 ml de ácido clorídrico 0,5 M e agitar. Adicionar 100 ml de metanol e submeter ao banho de ultra-som por 15 minutos e, em seguida, agitar mecanicamente por mais 15 minutos. Completar o volume com metanol e homogeneizar. Filtrar. Pesar, exatamente, cerca de 50 mg de glibenclamida padrão e transferir para balão volumétrico de 50 ml, dissolver em 35 ml de metanol e submeter ao banho de ultra-som por 15 minutos. Agitar mecanicamente por mais l5 minutos. Completar com metanol. Transferir 5 ml desta solução para balão volumétrico de 50 ml, adicionar 1 ml de ácido clorídrico 0,5 M e agitar. Completar o volume com metanol e homogeneizar. Medir as absorvâncias das soluções resultantes em 300 nm (V.2.14.-3) utilizando mistura de ácido clorídrico 0,5 M e metanol (1:49) para ajuste do zero. Calcular o teor de C23H28ClN3O5S nos comprimidos a partir das leituras obtidas.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
154
HIDRÓXIDO DE MAGNÉSIO
Magnesii hydroxidum
Mg(OH)2 58,32 0756.06-7
Contém, no mínimo, 95,0 % e, no máximo, 100,5 % de Mg(OH)2, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó branco, fino, amorfo, inodoro.
Solubilidade. Praticamente insolúvel em água. Solúvel em soluções de ácidos diluídos.
IDENTIFICAÇÃO
A. A suspensão aquosa da amostra apresenta reação alcalina quando adicionada de fenolftaleína SI.
B. Dissolver cerca de 15 mg da amostra em 2 ml de ácido nítrico SR e neutralizar com solução diluída de hidróxido de sódio SR. Esta solução responde às reações do íon magnésio (V.3.1.1)
ENSAIOS DE PUREZA
Aspecto da solução. Dissolver 5 g da amostra na mistura de 50 ml deácido acético e 50 ml de água. Apenas uma leve efervescência deve ser observada. Ferver por 2 minutos e diluir para 100 ml com ácido acético diluído. Filtrar através de funil de vidro sinterizado, previamente calcinado e tarado. A solução é límpida e incolo.
Substâncias solúveis. Misturar 2 g da amostra com 100 ml de água e ferver por 5 minutos. Filtrar ainda quente, através de funil de vidro sinterizado, deixar esfriar e diluir para 100 ml com água. Evaporar 50 ml do filtrado à secura e dessecar entre 100 oC e 105 oC. O resíduo não pesa mais do que 20 mg. No máximo 2%.
Substâncias insolúveis em ácido acético. Qualquer resíduo obtido em Aspecto da solução, após lavado com ácido acético diluído e calcinado a 600 oC, não pesa mais do que 5 mg. No máximo 1%.
Cloreto (V.3.2.1). Utilizar 5 ml da solução obtida em Aspecto da solução e proceder conforme descrito em Ensaio-limite para cloretos.
No máximo 0,1% (1 000 ppm).
Sulfato (V.3.2.2). Utilizar 2 ml da solução obtida em Aspecto da solução e proceder conforme descrito em Ensaio-limite para sulfatos.
No máximo 0,5% (5 000 ppm).
Arsênio (V.3.2.5). Utilizar 5 ml da solução obtida em Aspecto da solução e proceder conforme descrito em Ensaio-limite para arsênio.
No máximo 0,0004% (4 ppm).
Cálcio (V.3.2.7). Diluir 1,3 ml da solução descrita em Aspecto da solução para 150 ml com água. Prosseguir conforme descrito em Ensaio-limite para cálcio, utilizando 15 ml da solução amostra. No máximo 1,5%.
Metais pesados (V.3.2.3-2- Método I). Dissolver 1 g da amostra em 5 ml de ácido clorídrico diluído e agitar com 25 ml de metilisobutilcetona.
Deixar em repouso, separar a camada aquosa e evaporar até
secura. Dissolver o resíduo em 15 ml de água. Prosseguir conforme
descrito em Ensaio-limite para metais pesados utilizando 12 ml da
solução anterior. Preparar o padrão utilizando solução padrão de
chumbo (2 ppm ). No máximo 0,003% (30 ppm).
Ferro (V.3.2.4). Dissolver 0,15 g da amostra em 5 ml de ácido clorídrico diluído e diluir para 10 ml com água destilada. Utilizar 5 ml e proceder conforme descrito em Ensaio-limite para ferro. No máximo 0,07% (700 ppm).
Perda por dessecação (V.2.9). Determinar em estufa a 105 oC, por 2 horas. No máximo 2%.
Cinzas sulfatadas (V.2.10). Determinar em 0,5 g da amostra. Aquecer, gradativamente, até 900 oC e calcinar até peso constante. De 30,0% a 32,5%.
DOSEAMENTO
Dissolver 0,1 g da amostra em 2 ml de ácido clorídrico 2 M e proceder conforme descrito para magnésio em Titulações complexométricas (V.3.4.4). Cada ml de edetato dissódico 0,1 M SV equivale a 5,832 mg de Mg(OH)2.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CATEGORIA TERAPÊUTICA
Antiácido.
155
IBUPROFENO
Ibuprofenum
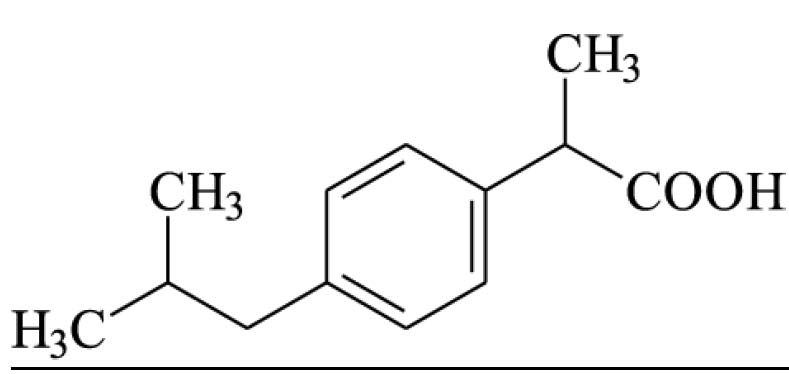
C13H18O2 206,28 0687.01-4
Ácido (±)-α-metil-4-(2-metilpropil)benzenoacético
Contém, no mínimo, 97,0% e, no máximo, 103,0% de C13H18O2, em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Pó cristalino branco ou quase branco, odor característico.
Solubilidade. Praticamente insolúvel em água, facilmente solúvel em etanol, acetona, metanol e clorofórmio, ligeiramente solúvel em acetato de etila. Solúvel em soluções aquosas de hidróxidos alcalinos.
Constantes físico-químicas
Faixa de fusão (V.2.2): 75 oC a 78 oC.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) de uma dispersão em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de ibuprofeno padrão, preparado de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 240 a 300 nm, de uma solução a 0,025% (p/V) em hidróxido de sódio 0,1 M, exibe máximos e mínimos somente nos mesmos comprimentos de onda de solução similar de ibuprofeno padrão. As respectivas absorvâncias, calculadas em relação à substância anidra, nos comprimentos de onda de 264 e 273 nm, não diferem mais que 3%.
C. Proceder conforme descrito em Substâncias relacionadas. A mancha principal obtida no cromatograma com a solução (3) corresponde em tamanho, cor e posição à mancha obtida no cromatograma com a solução (4).
ENSAIOS DE PUREZA
Limpidez da solução (IV.-3). A solução a 10% (p/V) em etanol é límpida.
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel G, como suporte, e mistura de n-hexano-acetato de etila-ácido acético glacial (15:5:1), como fase móvel. Aplicar, separadamente, à placa 5 μl de cada uma das soluções descritas a seguir, recentemente preparadas, em diclorometano.
Solução (1): solução a 10% (p/V) da amostra.
Solução (2): solução a 0,1% (p/V) da amostra.
Solução (3): solução a 0,5% (p/V) da amostra.
Solução (4): solução a 0,5% (p/V) de ibuprofeno padrão.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar, pulverizar com solução de p-dimetilaminobenzaldeído, aquecer em estufa a 100 oC por 5 minutos e pulverizar com solução aquosa de cloreto férrico a 5% (p/V). Nenhuma mancha secundária obtida no cromatograma com a solução (1) é mais intensa que a mancha obtida com a solução (2).
Poder rotatório (V.2.8). + 0,05o a - 0,05o, determinado em solução a 2,5% (p/V) em metanol.
Metais pesados (V.3.2.3 - Método II). No máximo 0,002% (20 ppm).
Água (V.2.20.1). No máximo 1%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g da substância. No máximo 0,5%.
DOSEAMENTO
Pesar, exatamente, cerca de 0,5 g da amostra e dissolver em 100 ml de etanol. Adicionar seis gotas de solução etanólica de fenolftaleína a 1% (p/V) e titular com hidróxido de sódio 0,1 M SV até viragem para rosa. Realizar ensaio em branco e fazer as correções necessárias.
Cada ml de hidróxido de sódio 0,1 M SV equivale a 20,628 mg de C13H18O2.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Analgésico, antiinflamatório.
________________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Solução de p-dimetilaminobenzaldeído
Preparação - Dissolver 1 g de p-dimetilaminobenzaldeído em 100 ml de etanol e adicionar 10 ml de ácido clorídrico
155.1
IBUPROFENO COMPRIMIDOS
Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C13H18O2.
IDENTIFICAÇÃO
A. Pesar e pulverizar os comprimidos. Extrair quantidade de pó equivalente a 0,5 g de ibuprofeno com 20 ml de acetona, filtrar e evaporar o filtrado até secura em corrente de ar, sem aquecimento. O espectro de absorção no infravermelho (V.2.14.-4) do resíduo, disperso em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de ibuprofeno padrão, preparado de maneira idêntica.
B. Recristalizar o resíduo obtido no teste A de Identificação com éter de petróleo (ponto de ebulição de 40 oC a 60 oC). O ponto de fusão
(V.2.2) é cerca de 75 oC.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO
Meio de dissolução: tampão fosfato pH 7,2, 900 ml
Aparelhagem: cesta, 150 rpm
Tempo: 30 minutos
Procedimento: após o teste, retirar alíquota do meio de dissolução e diluir em tampão fosfato pH 7,2 até concentração adequada. Medir as absorvâncias das soluções em 221 nm (V.2.14.-3), utilizando o mesmo solvente para ajuste do zero. Calcular a quantidade de C13H18O2 dissolvido no meio, comparando as leituras obtidas com a da solução de ibuprofeno padrão na concentração de 0,002% (p/V), preparada no mesmo solvente.
Tolerância: não menos que 70% (T) da quantidade declarada de C13H18O2 se dissolvem em 30 minutos.
ENSAIOS DE PUREZA
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel G, como suporte, e mistura de n-hexano-acetato de etila-ácido acético glacial (15:5:1), como fase móvel. Aplicar separadamente à placa 5 μl de cada uma das soluções descritas a seguir.
Solução (1): extrair quantidade de pó equivalente a 0,2 g de ibuprofeno
com três porções de 10 ml de clorofórmio, filtrar, evaporar
até cerca de 1 ml e adicionar quantidade suficiente de clorofórmio
para 2 ml.
Solução (2): diluir um volume da solução (1) para 100 volumes com clorofórmio.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar, pulverizar com solução de p-dimetilaminobenzaldeído, aquecer em estufa a 100 oC por 5 minutos e pulverizar com solução aquosa de cloreto férrico a 5% (p/V). Nenhuma mancha secundária obtida no cromatograma com a solução (1) é mais intensa que a mancha obtida com a solução (2).
Água (V.2.20.1). No máximo 5%.
DOSEAMENTO
Pesar e pulverizar 20 comprimidos. Extrair quantidade de pó equivalente a 0,5 g de ibuprofeno com 20 ml de clorofórmio. Filtrar em funil de vidro sinterizado e lavar o resíduo com três porções de 10 ml de clorofórmio. Evaporar o filtrado em banho de vapor. Dissolver o resíduo assim obtido em 50 ml de etanol, previamente neutralizado, com hidróxido de sódio 0,1 M SV, utilizando solução etanólica de fenolftaleína a 1% (p/V) como indicador. Titular com hidróxido de sódio 0,1 M SV até viragem para rosa. Cada ml de hidróxido de sódio 0,1 M SV equivale a 20,628 mg de C13H18O2.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
________________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Solução de p-dimetilaminobenzaldeído
Preparação - Dissolver 1 g de p-dimetilaminobenzaldeído em 100 ml de etanol e adicionar 10 ml de ácido clorídrico.
156
LANATOSÍDEO C
Lanatosidum C
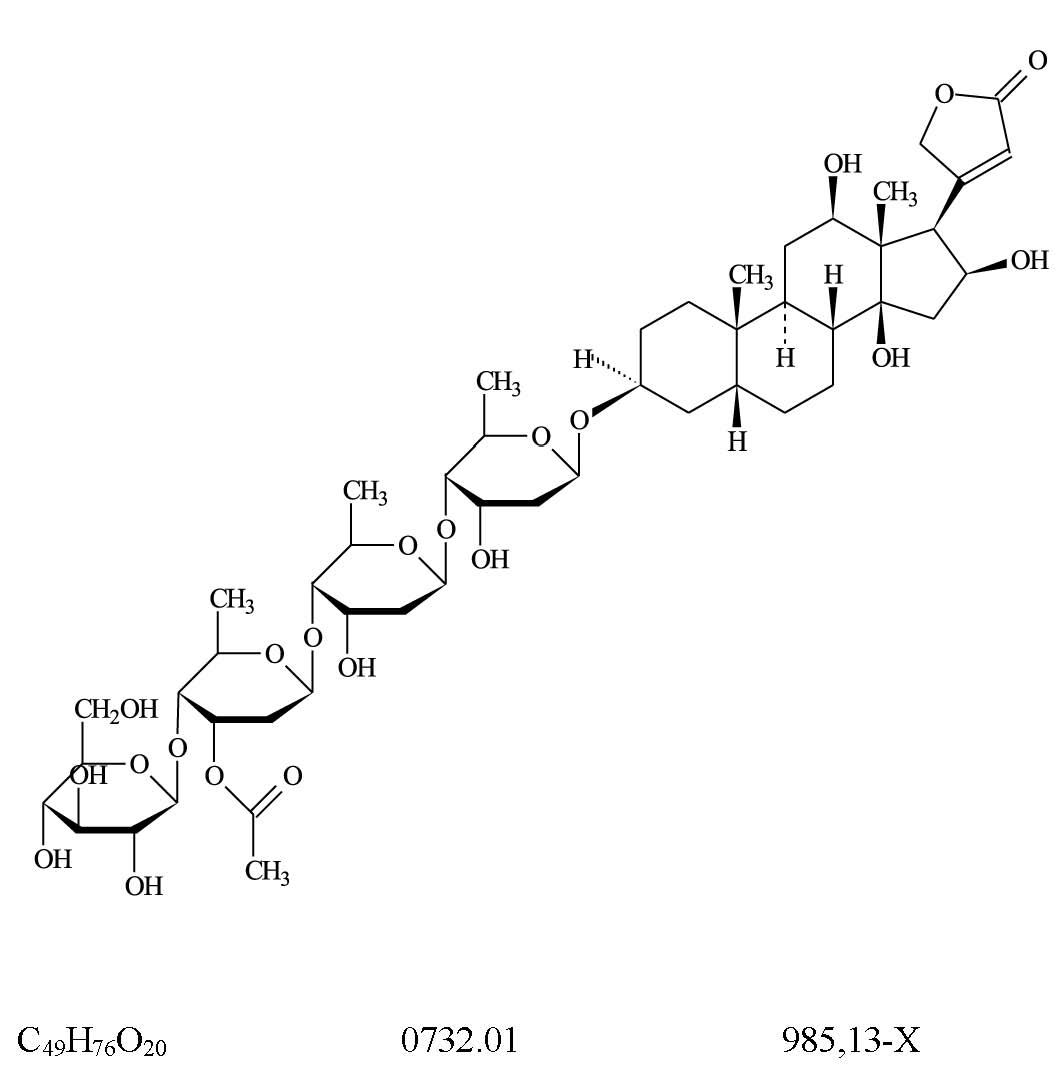
(3β,5β,12β?-[(O-β-D-glicopiranosil-(1→4)-O-(O-acetil-2,6-didesoxi- β-D-ribo-hexopiranosil)-(1→4)-O-2,6-didesoxi-β-D-ribo-exopiranosil-( 1→4)-2,6-didesoxi-β-D-ribo-hexopiranosil)oxi]-12,14-diidroxicard- 20(22)-enolídeo.
Contém, no mínimo, 97,0% e, no máximo, 102,0% de C49H76O20, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó cristalino, branco ou levemente amarelo, higroscópico e inodoro.
Solubilidade. Praticamente insolúvel em água, solúvel em metanol, dioxano e piridina anidra, insolúvel em clorofórmio e em éter etílico.
Constantes físico-químicas
Poder rotatório específico (V.2.8): + 32º a + 35,5º. Determinar, em solução a 2% (p/V) em metanol, em relação à substância dessecada.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) da amostra dessecada, até peso constante, e dispersa em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de lanatosídeo C padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Substâncias Relacionadas. A mancha principal obtida com a solução (2) corresponde em posição, cor e intensidade àquela obtida com a solução (3).
C. Dissolver 0,5 mg da amostra em 0,2 ml de etanol 60% (V/V) e adicionar 0,1 ml de solução etanólica a 2% (p/V) de ácido 3,5-
dinitrobenzóico e 0,1 ml de hidróxido de sódio 2 M. Desenvolve-se
coloração violeta.
D. Dissolver 5 mg da amostra em 5 ml de ácido acético glacial e adicionar 0,05 ml de cloreto férrico a 10,5% (p/V). Adicionar, cuidadosamente, sem agitar, 2 ml de ácido sulfúrico. Deixar em repouso.
Um anel castanho, não avermelhado, se desenvolve na interface e coloração verde-amarelada, que muda para azul-esverdeada, se difunde a partir do anel.
ENSAIOS DE PUREZA
Aspecto da solução. A solução a 2% (p/V) em metanol é límpida e incolor.
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel G, como suporte, e mistura de tolueno-etanol-cloreto de metileno-água (60:30:20:1), como fase móvel. Aplicar, separadamente, à placa 5 μl de cada uma das soluções, recentemente preparadas em metanol, descritas a seguir.
Solução (1): solução a 2% (p/V) da amostra.
Solução (2): solução a 0,2% (p/V) da amostra.
Solução (3): solução a 0,2% (p/V) de lanatosídeo C padrão.
Solução (4): solução a 0,03% (p/V) de lanatosídeo C padrão.
Solução (5): solução a 0,02% (p/V) de lanatosídeo C padrão.
Solução (6): solução a 0,01% (p/V) de lanatosídeo C padrão.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar, nebulizar com solução etanólica de ácido sulfúrico a 5% (V/V). No cromatograma obtido com a solução (1) nenhuma mancha secundáriaé mais intensa do que a mancha obtida no cromatograma com a solução (4), não mais que três manchas são mais intensas do que a mancha obtida no cromatograma com a solução (6), e não mais que uma destas manchas é mais intensa do que a obtida com a solução (5).
Perda por dessecação (V.2.9). Determinar em 0,5 g de amostra, em estufa à pressão reduzida a 105 ºC, com pentóxido fosforoso e pressão de 1,5 a 2,5 kPa, até peso constante. No máximo 7,5%.
Cinzas sulfatadas (V.2.10). Determinar em 0,1 g de amostra. No máximo 0,5%.
DOSEAMENTO
Pesar, exatamente, cerca de 50 mg de amostra e dissolver em etanol.
Completar o volume para 50 ml com o mesmo solvente. Diluir, sucessivamente, em etanol até concentração de 0,005% (p/V). Preparar solução padrão na mesma concentração, utilizando o mesmo solvente. A 5 ml de cada solução diluída adicionar 3 ml de picrato de sódio alcalino SR e deixar em repouso, em banho de água, na temperatura de 19 ºC a 21 ºC, por 40 minutos, ao abrigo da luz. Medir as absorvâncias das soluções amostra e padrão resultantes, em 484 nm (V.2.14.-3), utilizando mistura de 5 ml de etanol e 3 ml de solução de picrato de sódio alcalino SR para o ajuste do zero. Calcular o teor de C49H76O20 na amostra a partir das leituras obtidas.
EMBALAGEM E ARMAZENAMENTO
Em recipientes de vidro, bem-fechados, protegidos da luz e estocados em temperatura inferior a 10 ºC.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Glicosídeo cardiotônico.
157
LIDOCAÍNA
Lidocainum
2-(Dietilamino)-N-(2,6-dimetilfenil)acetamida
Contém, no mínimo, 99,0% e, no máximo, 101,0% de C14H22N2O, em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Pó cristalino branco ou quase branco.
Solubilidade. Praticamente insolúvel em água, muito solúvel em etanol e em cloreto de metileno, solúvel em éter etílico. Solúvel emácido clorídrico diluído.
Constantes físico-químicas
Faixa de fusão (V.2.2): 66 oC a 70 oC.
IDENTIFICAÇÃO
O teste de identificação B pode ser omitido se forem realizados os testes A, C, D e E. Os testes de identificação C, D e E podem ser omitidos se forem realizados os testes A e B.
A. Determinar a faixa de fusão na amostra não dessecada. O valor encontrado está na faixa de 66 oC a 70 oC.
B. O espectro de absorção no infravermelho (V.2.14.-4) da amostra dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de lidocaína padrão, preparado de maneira idêntica.
C. Dissolver, aquecendo, 0,2 g da amostra em mistura de 0,5 ml de ácido clorídrico diluído e 10 ml de água. Adicionar 10 ml de solução de ácido pícrico a 1% (p/V). O precipitado lavado com água e dessecado apresenta temperatura de fusão (V.2.2) ao redor de 230 ºC, com decomposição.
D. A cerca de 5 mg da amostra adicionar 0,5 ml de ácido nítrico fumegante. Evaporar até secura em banho-maria, esfriar e dissolver o resíduo em 5 ml de acetona. Adicionar 0,2 ml de hidróxido de potássio alcoólico 0,5 M. Desenvolve-se coloração verde.
E. Dissolver cerca de 0,1 g da amostra em 1 ml de etanol e adicionar 0,5 ml de solução a 10% (p/V) de nitrato de cobalto. Forma-se precipitado verde-azulado.
ENSAIOS DE PUREZA
Aspecto da solução. Dissolver 1 g da amostra em 3 ml de ácido clorídrico diluído e diluir para 10 ml com água. A solução é límpida e incolor.
2,6 dimetilanilina. Dissolver 0,25 g da amostra em metanol e diluir
para 10 ml com o mesmo solvente. A 2 ml da solução anterior,
adicionar 1 ml de solução a 1% (p/V) de dimetilaminobenzaldeído em
metanol e 2 ml de ácido acético glacial. Deixar em repouso por 10
minutos. Qualquer coloração amarela na solução em exame não é
mais intensa do que a de uma solução referência, preparada simultaneamente,
utilizando 2 ml de solução metanólica de 2,6 dimetilanilina
a 0,25% (p/V).
Cloreto (V.3.2.1). Dissolver 1,4 g da amostra em mistura de 3 ml deácido nítrico e 12 ml de água e proceder conforme descrito em Ensaio-limite para cloretos. No máximo 0,0035% (35 ppm).
Sulfato (V.3.2.2). Dissolver 0,5 g da amostra em 5 ml de etanol e diluir para 25 ml com água. Proceder conforme descrito em Ensaiolimite para sulfatos, utilizando 20 ml da solução. No máximo 0,1% (1 000 ppm).
Metais pesados (V.3.2.3 - Método II). Proceder conforme descrito em Ensaio-limite para metais pesados, utilizando 1 g da amostra. No máximo 0,002% (20 ppm).
Água (V.2.20.1). Determinar em 1,0 g da amostra. No máximo 0,1 %.
DOSEAMENTO
Proceder conforme descrito em titulação em meio não-aquoso (V.3.4.5). Pesar, exatamente, cerca de 0,2 g da amostra, adicionar 50 ml de ácido acético glacial e agitar até completa dissolução. Titular com ácido perclórico 0,1 M SV, determinando o ponto final potenciometricamente.
Cada ml de ácido perclórico 0,1 M equivale a 23,43 mg de C14H22N2O.
EMBALAGEM E ARMAZENAMENTO:
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Anestésico local.
________________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Picrato de sódio alcalino SR
Preparação - A 10 ml de solução de hidróxido de sódio a 5% (p/V), adicionar 20 ml de solução de ácido pícrico a 1% (p/V) e completar o volume para 100 ml com água.
158
MACELA
Achyroclines flos
Achyrocline satureioides (Lam.) DC. - ASTERACEAE
A droga vegetal consiste das sumidades floridas secas de Achyrocline satureioides (Lam.) DC. É permitida a presença de pedúnculos e pedicelos das inflorescências, em um comprimento de até 3 cm e correspondendo a um valor não maior do que 1% do peso seco do conjunto. A droga deve conter, no mínimo, 0,4% de óleo essencial e, no mínimo 1,7% de flavonóides totais calculados como quercetina (C15H10O7). E, no mínimo, 0,14 % de quercetina e 0,07% de luteolina.
SINONÍMIA CIENTÍFICA
Gnaphalium satureioides Lam.
SINONÍMIA VULGAR
Marcela, macela-do-campo.
CARACTERES ORGANOLÉPTICOS
Cor amarelo ouro, odor aromático e agradável, sabor levemente amargo.
A cor das inflorescências secas pode variar, não correspondendo a estágios de maturação.
DESCRIÇÃO MACROSCÓPICA
A droga, constituída pelos ramos com inflorescências, deve estar acompanhada de alguns ramos superiores não alados, para comprovar a identidade da espécie. Flores reunidas em capítulos agrupados em glomérulos, sendo estes por sua vez organizados em cimas paniculiformes.
Cada capítulo apresenta 4 a 8 flores dimorfas, protegidas por um invólucro subcilíndrico, de 0,40 cm a 0,60 cm de altura, formado por 9 a 14 brácteas involucrais imbricadas, amarelas, amareladas, amarelo-palha, amarelo-pálido a esverdeadas, ou ainda amarelo- douradas, amarelo-pardo a amarelo-avermelhadas. As brácteas involucrais são escariosas, hialinas, engrossadas na metade inferior, ao longo da nervura mediana, e estão dispostas em 3 ou 4 séries, sendo as séries exteriores gradualmente menores. Cada bráctea apresenta forma navicular, ápice acuminado e base truncada e mede aproximadamente 0,10 cm de largura. As brácteas mais internas são lanceolado- agudas, medindo 0,30 cm a 0,70 cm de comprimento e apresentando tricomas glandulares apenas na porção basal da face abaxial.
As demais brácteas são oblongas ou agudas, sendo que as medianas medem 0,35 cm a 0,45 cm de comprimento e as mais externas 0,25 cm a 0,30 cm de comprimento, apresentando ambas tricomas simples, lanosos, com 0,20 cm a 0,30 cm de comprimento e alguns tricomas glandulares na porção basal da face abaxial. O receptáculo é plano, alveolado, sem páleas. As flores marginais do capítulo, em número de 3 a 6, são pistiladas, com corola de 0,30 cm a 0,45 cm de comprimento, filiforme, às vezes dilatada na base, dentada ou partida no ápice, com alguns poucos tricomas glandulares na porção apical abaxial.
O estilete é filiforme, bífido, glabro, dilatado próximo à base, com ramos estigmáticos geralmente exsertos na maturação, de ápice truncado, papiloso e com uma coroa de tricomas na porção apical. O ovário é ínfero, bicarpelar e unilocular, com um único rudimento seminal, glabro, ovalado, levemente comprimido. O papus é unisseriado, com cerca de 20 cerdas brancas, ásperas, livres entre si na base, que alcançam quase a mesma altura da corola, raramente mais.
As flores do disco são em número de 1 a 3, hermafroditas, com corola tubulosa, estreita, de 0,30 cm a 0,45 cm de comprimento, de tubo ligeiramente dilatado na base e limbo pentadentado ou pentalobulado, dentes ou lóbulos com tricomas glandulares na face abaxial.
Androceu com 5 estames, epipétalos, inseridos na metade inferior da corola, com anteras sinânteras, de 0,15 cm a 0,20 cm de comprimento, com deiscência longitudinal e introrsa, sagitadas na base, com cauda laciniada; conetivo prolongado em um apêndice apical triangular, levemente obtuso, hialino. O ovário, o estilete e o papus são semelhantes aos das flores apenas pistiladas. Fruto do tipo aquênio, pardo, de 0,07 cm a 0,08 cm de comprimento, elipsoidal a obovado, levemente comprimido, glabro, de superfície papilosa.
DESCRIÇÃO MICROSCÓPICA
A face abaxial das brácteas apresenta epiderme formada por células alongadas, de contorno retangular, com paredes ligeiramente sinuosas, lisas, com tricomas tectores pluricelulares e unisseriados apenas no seu terço inferior. Ocorrem ainda tricomas glandulares, formados por um pedicelo bi a trisseriado, com 3 ou 4 camadas de células e por duas células terminais ovalado-alongadas, bem maiores do que as anteriores. Os tricomas glandulares das brácteas medem 60 μm a 100μm de comprimento total e sua cabeça possui diâmetro de 30 μm a 40 μm. As brácteas, em secção transversal, mostram poucas camadas de células junto à base, chegando a apenas duas camadas na porção mais apical. Em secção transversal, estas células são arredondadas, de paredes ligeiramente espessadas. O papus é constituído por cerdas longas, formadas por células alongadas, hialinas, de paredes finas, muitas delas projetadas lateralmente. A face abaxial da corola apresenta epiderme formada por células alongadas, de contorno poligonal.
Cinco feixes vasculares percorrem longitudinalmente seu tubo. Em secção transversal, a corola apresenta epiderme de células retangulares e um parênquima formado por 2 ou 3 camadas de células de contorno arredondado e de paredes um pouco espessadas. As lacínias são cobertas abaxialmente por tricomas glandulares, os quais medem de 60 μm a 90 μm de comprimento total e 30 μm a 40 μm de diâmetro na cabeça. O androceu, em secção transversal, apresenta as anteras recobertas abaxialmente por 2 camadas de células bem distintas.
A mais externa corresponde à epiderme, com células pequenas, arredondadas e de paredes espessas; a camada subjacente é constituída de uma fileira de células grandes, ovalado-alongadas no sentido radial e também com paredes espessas. Abaixo, ocorrem os sacos polínicos, formados por um parênquima de células pequenas e de paredes finas. Junto a estas encontra-se a camada mecânica, que consta de células arredondadas, com espessamento nas paredes laterais e basal. No momento da deiscência da antera, os sacos polínicos formam uma só loja. Os grãos de pólen são esferoidais e tricolpados, medindo de 17 μm a 35 μm de diâmetro e com exina espinhosa. O ovário é recoberto por uma camada de células epidérmicas de contorno poligonal, com algumas expansões na parede periclinal externa. Abaixo ocorre um tecido parenquimático constituído por várias camadas de células, que após o completo desenvolvimento, reduzem-se a 3 ou 4. Internamente, encontra-se apenas um rudimento seminal anátropo, preenchendo totalmente a cavidade ovariana. O embrião, quando desenvolvido, é formado quase que exclusivamente pelos cotilédones, restando apenas algumas células do endosperma, aderidas ao tegumento da semente. O estilete, em secção transversal, é circular, mostrando, próximo à base, uma expansão globosa, constituída por numerosas células arredondadas, de paredes finas. Externamente ocorre uma camada de células pequenas, regulares em forma e em tamanho. Internamente existe um parênquima de células de paredes muito delgadas, em cuja parte central ocorre um feixe vascular. O fruto, quando maduro, apresenta um pericarpo formado por 3 ou 4 camadas de células. O fruto é castanho-claro ou pardo, devido à coloração da primeira camada de células abaixo da epiderme. As demais camadas de células não possuem coloração e encontram-se aderidas ao tegumento da semente. Este é suberificado e adjacente ao resto do endosperma, que ocorre apenas na região superior e inferior da semente. Os cotilédones são pouco espessos, com face longitudinal plana e dorsal convexa.
DESCRIÇÃO MICROSCÓPICA DO PÓ
O pó atende a todas as exigências estabelecidas para a espécie, menos os caracteres macroscópicos. São característicos: coloração amarela ou uma variante de amarelo; brácteas involucrais ou porções das mesmas como descrito acima; flores inteiras ou suas porções, conforme descrição acima; estames ou partes destes com anteras sagitadas na base e cauda laciniada; estilete bífido de base dilatada;cerdas do papus com projeções laterais; aquênios pardos, de 0,07 cm a 0,08 cm de comprimento, elipsoidais a obovados, glabros, de superfície papilosa.
IDENTIFICAÇÃO
A. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando cromatoplaca de celulose como fase estacionária e clorofórmio-ácido acético glacial-água (50:45:5) como fase móvel. Aplicar, separadamente, em forma de banda, 5-10 μl da solução amostra e 2-3 μl da solução de referência, preparados como descrito a seguir.
Solução amostra: aquecer sob refluxo 10 g da droga em 100 ml deágua destilada, durante uma hora. Transferir o extrato para funil de decantação e extrair. Filtrar o extrato obtido, prensando e lavando o marco resultante com água aquecida. Extrair duas vezes com 50 ml, mais quatro vezes com 25 ml de acetato de etila. Lavar duas vezes o extrato obtido com 50 ml de água. Reunir as fases orgânicas, secar com sulfato de sódio anidro. Lavar o papel de filtro e o sulfato de sódio com acetato de etila. Evaporar em evaporador rotatório. Retomar o resíduo em 15 ml de metanol e proceder a análise cromatográfica.
Solução de referência: dissolver 1 mg de cada um dos padrões (quercetina, 3-O-metil-quercetina, luteolina e ácido caféico) em 100 μl de metanol, separadamente.
Desenvolver o cromatograma em percurso de aproximadamente 10 cm. Secar ao ar, à temperatura ambiente. Examinar a cromatoplaca sob luz ultravioleta (354 nm). Nebulizar com solução etanólica a 1 %(p/V) de difenilborato de amino-etanol (Reagente Natural A). Adicionalmente nebulizar com solução etanólica a 5 % (p/V) de polietilenoglicol 400. O cromatograma deverá apresentar uma mancha de fluorescência amarelo-ouro na mesma altura que a obtida com a solução de referência (Rf aproximadamente 0,40) correspondente à quercetina, outra de Rf cerca de 0,60 (luteolina) de cor marrom-claro, uma mancha de Rf aproximandamente 0,80 de coloração marromclaro atribuída à 3-O-mtil-quercetina e uma quarta pouco acima de fluorecência azul (Rf aproximandamente 0,90) relativa ao ácido cafeico.
ENSAIOS DE PUREZA
Matéria estranha (V.4.2.2.). No máximo 2%.
Determinação de água (V.4.2.3.). No máximo 10%.
Cinzas totais (V.4.2.4.). No máximo 8%.
DOSEAMENTO
A. Proceder conforme descrito em Determinação de óleos essenciais (V.4.2.6.). Utilizar um balão de 2 000 ml contendo quantidade deágua suficiente para encobrir a droga vegetal. Utilizar 30 g de flores não contundidas e destilar por 5 horas. Após extração, proceder imediatamenteà determinação do óleo essencial.
B. Determinar o teor de flavonóides totais. Pesar exatamente cerca de 0,400 g da droga pulverizada (800 μm) e colocar em balão de fundo redondo de 100 ml. Acrescentar 1 ml de uma solução de hexametilenotetramina a 0,5 % (p/V), 20 ml de acetona e 2 ml de ácido clorídrico. Aquecer em banho-maria, sob refluxo, por 30 minutos.
Filtrar a mistura para balão volumétrico de 100 ml. Lavar o resíduo da droga e o algodão em balão de fundo redondo, com duas porções de 20 ml de acetona, aquecendo a fervura sob refluxo, por 10 minutos.
Após resfriamento à temperatura ambiente, filtrar as soluções para o balão volumétrico, completando-se o volume com acetona. Em funil de separação, tratar 20 ml da solução com 20 ml de água e após extrair com 15 ml de acetato de etila, repetindo-se por três vezes, com porções de 10 ml de acetato de etila. Reunir as fases de acetato de etila e lavá-las em funil de separação, com duas porções de 50 ml deágua, transferindo a seguir para balão volumétrico de 50 ml, completando-se o volume com acetato de etila (solução-mãe SM). Pipetar 10 ml desta solução, adicionar 1 ml do reagente de cloreto de alumínio, diluindo-se em balão volumétrico de 25 ml com solução metanólica de ácido acético a 5% (V/V). Preparar o branco diluindo 10 ml da SM para 25 ml em balão volumétrico com solução metanólica de ácido acético a 5% (V/V). Após 30 minutos, medir a absorvância da solução a 425 nm, em cubeta de 1 cm, utilizando o branco para ajuste do zero. Calcular a porcentagem de flavonóides totais segundo a fórmula:
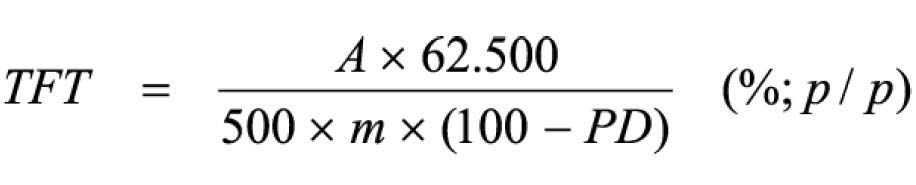
Em que
A = absorvância;
m = massa da droga (g);
PD = perda por dessecação (%; p/p)
O resultado é fornecido em porcentual (p/p) de flavonóides totais calculados como quercetina (C15H10O7).
C. Determinar o teor de quercetina e luteolina. Proceder conforme descrito em Cromatografia líquida de alta eficiência (V.2.17.4). Cerca de 18 g da droga seca e moída (800 μm), exatamente pesados, é extraída previamente, em aparelho tipo Soxhlet, com 300 ml de nhexano, durante 3 horas. O extrato é desprezado e o marco extraído com 300 ml de acetato de etila, durante 3 horas. O extrato obtido é evaporado à secura em evaporador rotatório e o resíduo retomado, quantitativamente, em metanol. A solução é transferida para balão volumétrico, ajustando-se o volume de 10 ml com o metanol. Uma alíquotade 1 ml desta solução é diluída para 50 ml, completando-se o volume com o solvente. Desta solução, uma alíquota de 4 ml é diluída volumetricamente a 20 ml utilizando como solvente uma mistura de metanol:água na proporção de 53:47. As amostras são filtradas através de filtro de membrana fluoreto de polivinilideno (0,45 μm de diâmetro nominal de poro) e injetadas no cromatógrafo.
As extrações e as injeções são realizadas em triplicata e o resultado é expresso pela média das determinações em gramas de quercetina e luteolina por 100 gramas da droga (%, p/p).
Curva de calibração: cerca de 5 mg das substâncias referência, quercetina e luteolina, são exatamente pesadas e dissolvidas em metanol.
A solução, com os dois flavonóides, é transferida para balão volumétrico de 100 ml, completando-se o volume com metanol (Solução- Mãe). Alíquotas da Solução-Mãe são diluídas com uma mistura metanol:água (53:47) obtendo-se soluções contendo quercetina e luteolina, nas seguintes concentrações: 1,5; 2,5; 5; 7,5; 10 μg/ml. As soluções são filtradas através de filtro de membrana de fluoreto de polivinilideno (0,45 μm de diâmetro nominal de poro) e injetadas no cromatógrafo.
Condições cromatográficas: a análise cromatográfica é realizada em cromatógrafo equipado com detector ultravioleta. As condições cromatográficas empregadas são: pré-coluna contendo sílica octadecilsililizada, 10 μm; coluna em aço inoxidável (250 mm x 4 mm d.i.) empacotada com sílica octadecilsililizada, 5 μm. A fase móvel é constituída de mistura de metanol:solução de ácido fosfórico 1 % (m/V) na proporção de 53:47; fluxo de 0,6 ml/min; detecção em 362 nm. A fase móvel é previamente filtrada através de membrana de fluoreto do polivinilideno 0,45 μm de diâmetro de poro.
Determinar a área do pico de quercetina e luteolina utilizando a Curva de Calibração.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, ao abrigo da luz e do calor, por um período não superior a um ano.
________________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Reagente de cloreto de alumínio
Dissolver 1g de cloreto de alumínio com solução metanólica de ácido acético 5% (V/V), em balão volumétrico de 500 ml.
Completar o volume.
159
MEBENDAZOL
Mebendazolum
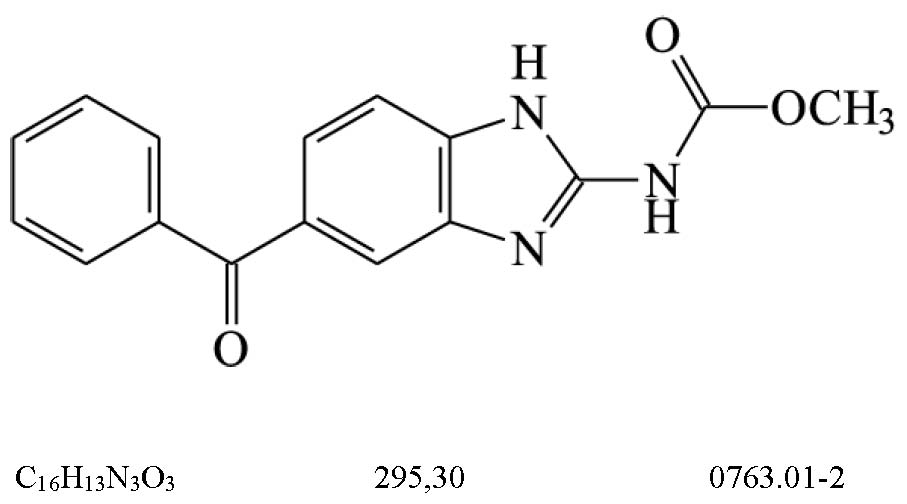
Éster metílico do ácido [5-benzoil -1H-2-benzimidazolil]carbâmico
Contém, no mínimo, 98,0% e, no máximo, 102,0% de C16H13N3O3, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó fino, amarelado e inodoro.
Solubilidade. Praticamente insolúvel em água, etanol, éter etílico e clorofórmio, solúvel em ácido fórmico.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14-4) da amostra dessecada a 105 ºC, até peso constante, e dispersa em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de mebendazol padrão, preparado de maneira idêntica.
B. Dissolver 30 mg da amostra em 2 ml de ácido fórmico e diluir, sucessivamente, em isopropanol até a concentração de 0,00075% (p/V). O espectro de absorção no ultravioleta (V.2.14-3), na faixa de 230 a 320 nm, desta solução, exibe máximos em 247 e 312 nm, idênticos aos observados no espectro de solução similar de padrão.
C. Dissolver 40 mg em 2 ml de ácido fórmico e adicionar 5 ml de etanol acidulado com algumas gotas de ácido clorídrico. Agitar vigorosamente e filtrar. Adicionar ao filtrado cerca de 3 mg de cloridrato de p-fenilenodiamina e agitar. Adicionar cerca de 0,1 g de zinco em pó e deixar em repouso por 2 minutos. Adicionar 5 ml de sulfato férrico amoniacal ácido SR. Produz-se coloração violeta.
ENSAIOS DE PUREZA
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de clorofórmio-metanol-ácido fórmico 96% (90:5:5), como fase móvel. Aplicar, separadamente, à placa, 10 μl de cada uma das soluções, recentemente preparadas, descritas a seguir.
Solução (1): dissolver 50 mg da amostra em 1 ml de ácido fórmico 96% e completar o volume para 10 ml, com clorofórmio.
Solução (2): dissolver 50 mg de mebendazol padrão em 1 ml de ácido fórmico 96% e completar o volume para 10 ml, com clorofórmio.
Solução (3): transferir 1 ml da solução (2) para balão volumétrico de 200 ml, completar o volume com mistura de clorofórmio e ácido fórmico 96% (9:1). Homogeneizar.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e examinar sob luz ultravioleta (254 nm). A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (2). Qualquer mancha secundária presente na solução (1) não deve ser mais intensa que a obtida com a solução (3).
Metais pesados (V.3.2.3 - Método II). No máximo 0,002% (20 ppm).
Perda por dessecação (V.2.9). Determinar em 1 g de amostra, em estufa a 105 ºC, por 4 horas. No máximo 0,5%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,1%.
DOSEAMENTO
Proceder conforme descrito em titulação em meio não-aquoso (V.3.4.5). Pesar, exatamente, cerca de 0,225 g da amostra e dissolver em 30 ml de ácido acético glacial. Titular com ácido perclórico 0,1 M SV determinando o ponto final potenciometricamente. Realizar ensaio em branco e fazer as correções necessárias. Cada ml de ácido perclórico 0,1 M equivale a 29,530 mg de C16H13N3O3.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Anti-helmíntico.
________________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Sulfato férrico amoniacal ácido SR
Preparação: dissolver 20 g de sulfato férrico amoniacal em 70 ml deágua, adicionar 10 ml de ácido sulfúrico 0,05 M e completar o
volume com água para 100 ml.
159.1
MEBENDAZOL SUSPENSÃO ORAL
Contém, no mínimo, 90,0% e, no máximo, 110,0%, da quantidade declarada de C16H13N3O3.
IDENTIFICAÇÃO
A. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 200 a 400 nm, da solução amostra preparada no método A de Doseamento exibe máximos e mínimos idênticos aos observados no espectro da solução padrão.
B. A mancha principal obtida com a solução (1) em Substâncias relacionadas corresponde em posição, cor e intensidade àquela obtida com a solução (2).
CARACTERÍSTICAS
Aspecto. Esvaziar completamente o conteúdo de 10 frascos, previamente agitados, em provetas correspondentes, limpas e secas, providas de tampa e observar imediatamente sob condições adequadas de visibilidade. O conteúdo deve escorrer com fluidez, a suspensão deve se apresentar homogênea, viscosa, livre de grumos e partículas estranhas.
Após 24 horas de repouso pode apresentar ligeira sedimentação, que deve ressuspender após agitação.
Determinação de volume (V.1.2). Cumpre o teste.
pH (V.2.19). 4,0 a 7,5.
ENSAIOS DE PUREZA
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de clorofórmio, metanol e ácido fórmico (90:5:5), como fase móvel. Aplicar separadamente à placa, 50 μl de cada uma das soluções, recentemente preparadas, descritas a seguir.
Solução (1): a uma alíquota equivalente a 10 mg de mebendazol adicionar 1 ml de ácido fórmico, agitar até dissolução, completar o volume para 10 ml com clorofórmio, homogeneizar e filtrar.
Solução (2): pesar 10 mg de mebendazol padrão, adicionar 1 ml deácido fórmico, agitar até dissolução, completar para 10 ml com clorofórmio e homogeneizar.
Solução (3): transferir 1 ml da solução (2) para um balão volumétrico de 200 ml, completar com mistura de clorofórmio e ácido fórmico (9:1) e homogeneizar.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e examinar sob luz ultravioleta (254 nm). Qualquer mancha obtida no cromatograma com a solução (1), diferente da mancha principal, não deve ser maior nem mais intensa que a mancha obtida com a solução (3).
TESTES DE SEGURANÇA BIOLÓGICA
Contagem de microrganismos viáveis (V.5.1.6.1). Bactérias aeróbicas totais: no máximo 1 000 UFC/ml. Fungos e leveduras: no máximo 100 UFC/ml.
Pesquisa e identificação de patógenos (V.5.1.7). Cumpre o teste.
DOSEAMENTO
A. Por espectrofotometria de absorção no ultravioleta (V.2.14-3).
Transferir volume da amostra equivalente a 100 mg de mebendazol
para balão volumétrico de 100 ml. Adicionar 30 ml de ácido fórmico
e agitar até completa dissolução. Completar o volume com ácido
fórmico e misturar. Transferir 5 ml desta solução para balão volumétrico
de 50 ml, adicionar 5 ml de ácido clorídrico 0,1 M, agitar
e completar o volume com isopropanol. Aquecer até leve fervura e
filtrar. Esfriar e diluir em isopropanol até a concentração de 0,01%
(p/V). Preparar solução padrão na mesma concentração, utilizando os
mesmos solventes. Medir as absorvâncias das soluções resultantes em
310 nm, utilizando ácido clorídrico 0,1 M e isopropanol (1:9) para o
ajuste do zero. Calcular o teor de C16H13N3O3 na amostra a partir das
leituras obtidas.
B. Por espectrofotometria de absorção no ultravioleta (V.2.14-3).
Transferir volume da amostra contendo o equivalente a 100 mg de mebendazol para balão volumétrico de 100 ml, adicionar 50 ml deácido fórmico e colocar em banho-maria a 50 ºC durante 15 minutos.
Esfriar, completar o volume com água, homogeneizar e filtrar através de um filtro de vidro de média porosidade. Transferir 10 ml do filtrado para um funil de separação, adicionar 50 ml de clorofórmio, 50 ml de água e agitar durante 2 minutos. Deixar separar as fases e transferir a camada clorofórmica para um segundo funil de separação.
Lavar a camada aquosa com duas porções de 10 ml de clorofórmio e adicionar os lavados clorofórmicos ao segundo funil de separação.
Lavar os extratos clorofórmicos combinados com uma mistura de ácido clorídrico M e ácido fórmico 10% (V/V) (4:50). Transferir a camada clorofórmica para um balão volumétrico de 100 ml. Extrair a camada aquosa com duas porções de 10 ml de clorofórmio, adicionar o extrato ao balão volumétrico, completar com isopropanol e misturar.
Diluir, sucessivamente, em isopropanol até a concentração de 0,005% (p/V). Preparar solução padrão na mesma concentração, utilizando os mesmos solventes. Preparar o branco como descrito a seguir. Transferir 45 ml de clorofórmio para um balão volumétrico de 100 ml, adicionar 1 ml de ácido fórmico 10%, completar com isopropanol, homogeneizar, transferir 5 ml desta solução para um balão volumétrico de 100 ml, completar com isoproanol e homogeneizar. Medir as absorvâncias das soluções resultantes em 274 nm, utilizando o branco para ajuste do zero. Calcular a quantidade de C16H13N3O3 na suspensão oral a partir das leituras obtidas.
EMBALAGEM E ARMAZENAMENTO
Em frascos de vidro âmbar, bem-fechados.
ROTULAGEM
Observar a legislação vigente.
160
MERBROMINA
Merbrominum
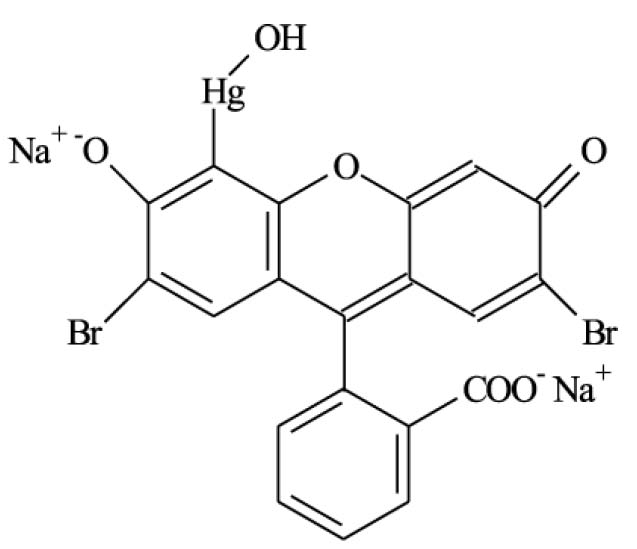
C20H8Br2HgNa2O6 750,70 0794.01-5
Sal dissódico, de (2',7'-dibromo-3',6'-diidroxi-3-oxospiro[isobenzofurano- 1 (3H), 9'-[9H)xanteno]-4'-il) hidroximercurio Contém, no mínimo, 22,4% e, no máximo, 26,7% de mercúrio (Hg = 200,59) e, no mínimo, 18,0% e, no máximo, 22,4% de bromo (Br = 79,9), calculados em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Escama ou grânulo verde-metálico a castanhoavermelhado.
Solubilidade. Facilmente solúvel em água, porém, algumas vezes deixa pequena quantidade de matérias insolúveis, praticamente insolúvel em etanol, acetona, éter etílico e em clorofórmio.
IDENTIFICAÇÃO
A. Uma solução 1:2 000 apresenta cor vermelha e fluorescência verde amarelada.
B. A 5 ml de uma solução 1:250 adicionar 3 gotas de ácido sulfúrico 10 % (p/V). Forma-se precipitado laranja-avermelhado.
C. Aquecer 0,1 g da amostra com pequenos cristais de iodo em tubo de ensaio; cristais vermelhos são sublimados na parte superior do tubo. Se forem produzidos cristais amarelos, atritar com bastão de vidro; a cor dos cristais passa para vermelho.
D. Pesar 0,1 g da amostra e adicionar 12 ml de solução de hidróxido de sódio 1:6. Evaporar até secura com agitação e incinerar a 600 ºC por 1 hora. Dissolver o resíduo em 5 ml de água e acidificar comácido clorídrico. Adicionar 3 gotas de solução saturada de cloro, 2 ml de clorofórmio e agitar; na camada clorofórmica produz-se cor castanho- amarelada.
ENSAIOS DE PUREZA
Cor de líquidos (V.2.12). Dissolver 0,4 g em 20 ml de água, adicionar 3 ml de ácido sulfúrico 10% (p/V) e filtrar. A cor do filtrado é menos intensa que a da solução padrão de cor C.
Halogênios solúveis
Preparação da amostra: dissolver 5 g da amostra em 80 ml de água,adicionar 10 ml de ácido nítrico 10 % (p/V) e completar o volume com água para 100 ml. Homogeneizar e filtrar. Transferir 40 ml do filtrado para tubo de Nessler, adicionar 6 ml de ácido nítrico 10 % (p/V) e completar para 50 ml com água.
Preparação do controle: em tubo de Nessler adicionar 0,25 ml deácido clorídrico 0,01 M, 6 ml de ácido nítrico 10% (p/V) e completar para 50 ml com água.
Procedimento: adicionar aos tubos 1 ml de nitrato de prata 0,1 M, misturar bem e deixar em repouso por 5 minutos ao abrigo da luz. A preparação amostra não produz turvação mais intensa que a produzida pela preparação controle.
Sais de mercúrio solúveis
Preparação da amostra: transferir para um tubo de ensaio, 5 ml do filtrado obtido no ensaio cor de líquidos e adicionar 5 ml de água.
Preparação do controle: dissolver 40 mg de cloreto de mercúrio II, exatamente pesados, em água e completar o volume para 1 000 ml. A 20 ml desta solução adicionar 3 ml de ácido sulfúrico 10 % (p/V).
Transferir para um tubo 5 ml da solução precedente e adicionar 5 ml de água.
Procedimento: adicionar aos tubos 1 gota de sulfeto de sódio SR. A cor da solução amostra não deve ser mais intensa que a da solução controle.
Compostos de mercúrio insolúveis. Dissolver 2,5 g da amostra em 50 ml de água e deixar em repouso por 24 horas ao abrigo da luz.
Centrifugar e lavar o precipitado com pequenas porções de água até que a última lavagem seja incolor. Transferir o precipitado para frasco com rolha esmerilhada, adicionar exatamente 5 ml de iodo 0,1 M SV e deixar repouso por 1 hora, agitando freqüentemente. Adicionar gota a gota, 4,3 ml de tiossulfato de sódio 0,1 M SV com agitação, usando 1 ml de amido SI. Produz-se coloração azul.
Perda por dessecação (V.2.9). Determinar em estufa a 105 ºC por 5 horas, em 2 g de amostra. No máximo 5%.
DOSEAMENTO
Mercúrio. Pesar, exatamente, cerca de 0,6 g de amostra previamente pulverizada e dessecada, colocar em vidraria apropriada com rolha esmerilhada e dissolver com 50 ml de água, acrescentar 8 ml de ácido acético glacial, 20 ml de clorofórmio e exatamente 30 ml de iodo 0,1 M SV. Tampar hermeticamente e deixar em repouso por 1 hora agitando, freqüentemente, com vigor. Titular o excesso de iodo com tiossulfato de sódio 0,1 M SV, com agitação vigorosa, usando 1 ml de amido SI. Realizar ensaio em branco e fazer as correções necessárias.
Cada ml de iodo 0,1 M SV equivale a 10,030 mg de Hg.
Bromo. Pesar, exatamente, em cadinho de porcelana, cerca de 0,5 g de amostra previamente pulverizada e dessecada, acrescentar 2 g de nitrato de potássio, 3 g de carbonato de potássio, 3 g de carbonato de sódio anidro e homogeneizar. Cobrir a superfície da mistura com 3 g de partes iguais de carbonato de potássio e carbonato de sódio anidro e calcinar entre 400 ºC e 500 ºC por 1 hora. Resfriar, dissolver e transferir quantitativamente a mistura calcinada para erlenmeyer, com o auxílio de 80 ml de água quente, e acidificar com ácido nítrico. Adicionar, exatamente, 25 ml de nitrato de prata 0,1 M e agitar. Titular o excesso de nitrato de prata com tiocianato de amônio 0,1 M SV usando 2 ml de sulfato férrico amoniacal SR como indicador. Realizar um ensaio em branco e fazer as correções necessárias. Cada ml de nitrato de prata 0,1 M equivale a 7,990 mg de Br.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados e opacos.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Conservante.
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Ácido sulfúrico 10% (p/V)
Especificação: contém 57 ml de ácido sulfúrico em água a 1 000 ml.
Conservação: em recipientes bem fechados.
Ácido nítrico 10% (p/V)
Especificação: contém 105 ml de ácido nítrico em água a 1 000 ml.
Conservação: em recipientes bem fechados ao abrigo da luz.
Solução saturada de Cloro
Preparação: preparar uma solução saturada de cloro em água.
Conservação: guardar a solução em frascos pequenos e completamente cheios, em local frio e escuro.
Observações: a solução tende a se deteriorar mesmo se protegida da luz e do ar. Para obter plena concentração preparar solução no momento do uso.
161
MESILATO DE PEFLOXACINO
Pefloxacin mesylate
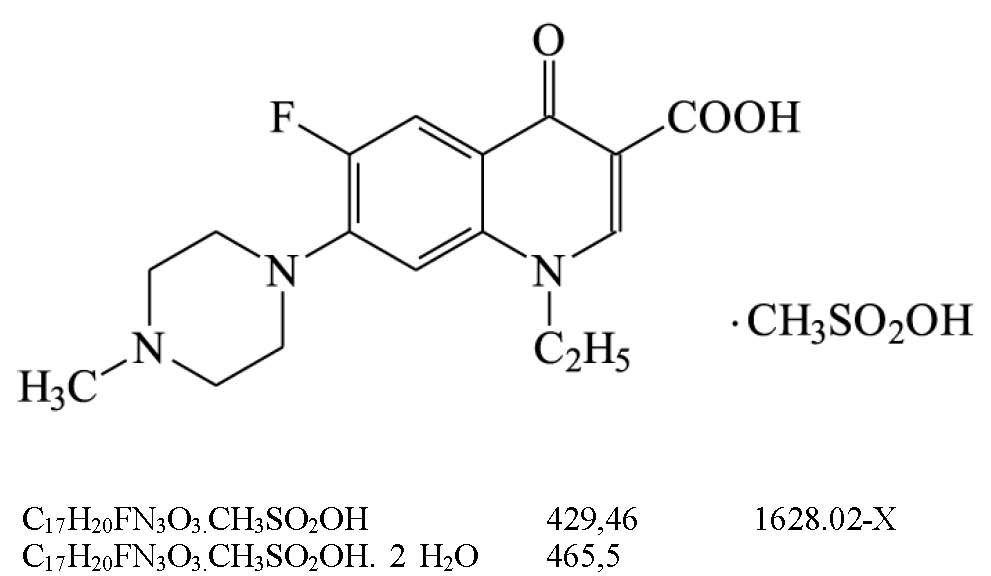
Metanosulfontato do ácido 1-etil-6-flúor-1,4-diidro-7-(4-metil-1-piperazinil)- 4-oxo-3-quinolino-carboxílico.
Contém, no mínimo, 98,5% e, no máximo, 101,5% de C17H20FN3O3CH3SO2OH, em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Pó amorfo branco.
Solubilidade. Muito solúvel em água. Pouco solúvel em etanol. Insolúvel em soluções ácidas.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) de amostra dessecada a 105ºC, até peso constante e dispersa em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de pefloxacino padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte e mistura de cloreto de metileno-metanol-hidróxido de amônio- acetonitrila (4:4:2:1), como fase móvel. Aplicar, separadamente, à placa, 10 μl de cada uma das soluções, recentemente preparadas, descritas a seguir.
Solução (1): solução a 1% (p/V) da amostra em metanol e água (1:1).
Solução (2): solução a 1% (p/V) de mesilato de pefloxacino padrão em metanol e água (1:1).
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e visualizar as manchas obtidas sob luz ultravioleta (254 nm e 336 nm).
A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (2).
ENSAIOS DE PUREZA
Água (V.2.20.1). Entre 7,7% e 10,9%.
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17) pelo método de difusão em ágar, utilizando cilindros.
Microrganismo: Staphylococcus epidermidis ATCC 12228.
Meio de cultura: número 1 de para manutenção do microrganismo;
solução salina estéril, para padronização do inóculo e meio número 11 para a camada base e camada de inóculo na placa.
Solução amostra: pesar, exatamente, cerca de 2 g de amostra, transferir, quantitativamente, para balão volumétrico de 500 ml. Adicionar 400 ml de água e agitar por 30 minutos. Completar o volume comágua e filtrar. Diluir, sucessivamente, até as concentrações de 8μg/ml, 16 μg /ml e 32 μg/ml, utilizando tampão fosfato de potássio 0,1 M, pH 8,0, estéril (Solução 2) como diluente.
Solução padrão: pesar, exatamente, cerca de 50 mg de mesilato de pefloxacino padrão e transferir quantitativamente para balão volumétrico de 50 ml utilizando água como solvente. Diluir, sucessivamente, até as concentrações de 8 μg /ml, 16 μg /ml e 32 μg /ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) como diluente.
Procedimento: adicionar 20 ml de meio de cultura número 11 em cada placa, esperar solidificar e adicionar 5 ml de inóculo a 1,0% e proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17.1), adicionando aos cilindros, 0,2 ml das soluções recentemente preparadas.
DOSEAMENTO
A. Por espectrofotometria de absorção no ultravioleta (V.2.14.-3).
Pesar, exatamente, cerca de 2 g de amostra, transferir com auxílio de 400 ml de água para balão volumétrico de 500 ml. Agitar por 30 minutos, completar o volume com o mesmo solvente e filtrar. Diluir, sucessivamente, até a concentração de 0,0006% (p/V), utilizandoágua como solvente. Preparar a solução padrão na mesma concentração, utilizando o mesmo solvente. Medir as absorvâncias das soluções resultantes em 276 nm, utilizando água para o ajuste do zero.
Calcular o teor de C17H20FN3O3.CH3SO2OH na amostra, a partir das leituras obtidas.
B. Por Cromatografia líquida de alta eficiência (V.2.17.4). Utilizar cromatógrafo líquido provido de detector ultravioleta a 276 nm; coluna
de 150 mm de comprimento e 3,9 de diâmetro interno, empacotada
com sílica quimicamente ligada a octadecilsilano; fluxo de 1
ml/minuto.
Fase móvel: ácido fosfórico 0,025 M com pH ajustado para 3,0com trietilamina-acetonitrila (87:15);
Solução amostra: pesar, exatamente, cerca de 2 g de amostra e transferir para balão volumétrico de 500 ml com auxílio de 400 ml deágua. Agitar por 30 minutos, completar o volume com água e filtrar. Diluir, sucessivamente, até concentração de 0,06 mg/ml, utilizandoágua como solvente.
Solução padrão: pesar, exatamente, cerca de 50 mg de mesilato de pefloxacino padrão, transferir para balão volumétrico de 50 ml e completar o volume com água. Diluir, sucessivamente, até concentração de 0,06 mg/ml, utilizando água como solvente.
A eficiência da coluna não deve ser menor do que 10 000 pratos teóricos/metro. O desvio padrão relativo das áreas de replicatas dos picos registrados não deve ser maior que 2%.
Procedimento: injetar separadamente 20 μl das soluções amostra e padrão, registrar os cromatogramas e medir as áreas dos picos. Calcular teor de C17H20FN3O3.CH3SO2OH na amostra a partir das respostas obtidas para as soluções padrão e solução amostra.
EMBALAGEM E ARMAZENAMENTO:
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antimicrobiano.
161.1
MESILATO DE PEFLOXACINO COMPRIMIDOS
Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C17H20FN3O3.
IDENTIFICAÇÃO
A. Pesar e pulverizar os comprimidos. Pesar do pó o equivalente a 0,1 g de pefloxacino, adicionar 15 ml de água e agitar. Completar o volume para 25 ml e filtrar. Adicionar ao filtrado 10 ml de metanol e evaporar em rotavapor, à temperatura de 30 °C. Deixar o resíduo em dessecador, até peso constante. O espectro de absorção no infravermelho (V.2.14.-4) do resíduo disperso em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de mesilato de pefloxacino padrão, preparado de maneira idêntica.
B. Proceder conforme descrito no teste B de Identificação na monografia de mesilato de pefloxacino. Preparar a solução (1) e solução (2) como descrito a seguir.
Solução (1): pesar e pulverizar os comprimidos. Transferir quantidade do pó equivalente a 0,25 g de pefloxacino para balão volumétrico de 25 ml, adicionar mistura de metanol e água (1:1), agitar, completar o volume com o mesmo solvente e filtrar.
Solução (2): utilizar mesilato de pefloxacino padrão, para preparar solução a 1% (p/V) em pefloxacino, utilizando mistura de metanol eágua (1:1) como solvente.
CARACTERÍSTICAS
Determinação do peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito em Determinação da potência na monografia de mesilato de pefloxacino. Preparar a solução amostra e solução padrão como descrito a seguir.
Solução amostra: pesar e pulverizar 20 comprimidos. Transferir quantidade de pó equivalente a 2 g de pefloxacino para balão volumétrico de 500 ml com auxilio de 400 ml de água. Agitar por 30 minutos, completar o volume com água e filtrar. Diluir, sucessivamente, até as concentrações de 8 g/ml, 16 g/ml e 32 μg/ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) como diluente.
Solução padrão: pesar, de mesilato de pefloxacino padrão, o equivalente a cerca de 50 mg de pefloxacino e transferir quantitativamente para balão volumétrico de 50 ml utilizando água como solvente.
Diluir, sucessivamente, até as concentrações de 8 μg/ml, 16μg/ml e 32 μg/ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (solução 2) como diluente.
DOSEAMENTO
A. Por espectrofotometria de absorção no ultravioleta. Pesar e pulverizar 20 comprimidos. Utilizar quantidade de pó equivalente a 2 g de pefloxacino, transferir com auxílio de 400 ml de água para balão volumétrico de 500 ml. Agitar por 30 minutos, completar o volume com o mesmo solvente e filtrar. Diluir sucessivamente, até a concentração de 0,0006% (p/V), utilizando água como solvente. Preparar a solução padrão de mesilato de pefloxacino na mesma concentração em pefloxacino, utilizando água como solvente. Medir as absorvâncias das soluções resultantes em 276 nm (V.2.14.-3), utilizando água para o ajuste do zero. Calcular a quantidade de C17H20FN3O3 nos comprimidos, a partir das leituras obtidas.
B. Por cromatografia líquida de alta eficiência (V.2.17.4). Proceder conforme descrito no método B de Doseamento na monografia de mesilato de pefloxacino. Preparar a solução amostra e solução padrão como descrito a seguir.
Solução amostra: pesar e pulverizar 20 comprimidos. Utilizar quantidade do pó equivalente a 2 g de pefloxacino e transferir para balão volumétrico de 500 ml com auxílio de 400 ml de água. Agitar por 30 minutos, completar o volume com água e filtrar. Diluir, sucessivamente até concentração de 0,6 mg/ml, utilizando água como diluente.
Solução padrão: pesar, em mesilato de pefloxacino padrão, o equivalente a cerca de 50 mg de pefloxacino e transferir quantitativamente para balão volumétrico de 50 ml utilizando água como solvente.
Diluir, sucessivamente, até a concentraçãode 0,05 mg/ml, utilizando água como solvente.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
162
METILPARABENO
Methylis parahidroxibenzoas
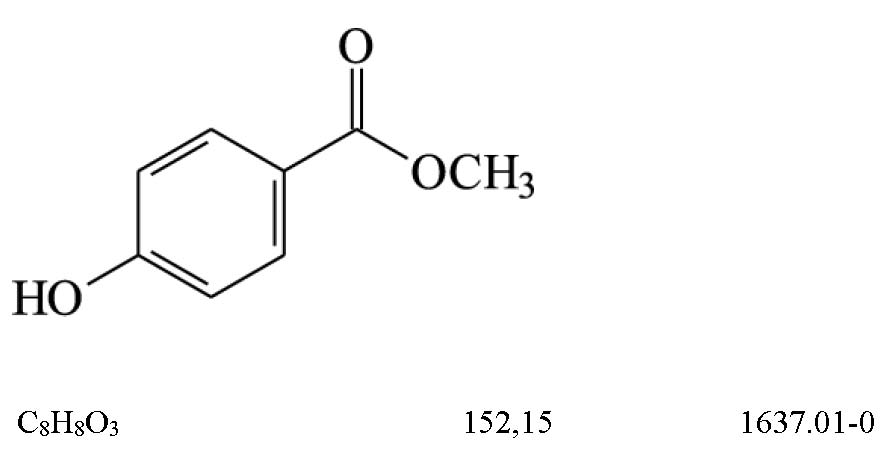
Éster metílico do ácido 4-hidroxibenzóico
Contém, no mínimo, 99,0% e, no máximo, 101,0% de C8H8O3.
DESCRIÇÃO
Caracteres físicos. Pó cristalino branco ou cristais incolores.
Solubilidade. Muito pouco solúvel em água, solúvel em álcool, éter e metanol.
Constantes físico-químicas
Faixa de fusão (V.2.2): entre 125 oC e 128 oC.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14) da amostra, dispersa em brometo de potássio, apresenta máximos de absorção nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de metilparabeno padrão, preparado de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 230 a 280 nm, de uma solução a 0,0005% (p/V) em etanol, exibe máximo em 258 nm e a absorvância é de 0,52 a 0,56.
ENSAIOS DE PUREZA
Aspecto da solução (IV.-3). A solução a 10% (p/V) em etanol é límpida e incolor.
Acidez. A 2 ml da solução obtida no ensaio Aspecto da solução adicionar 3 ml de etanol, 5 ml de água isenta de dióxido de carbono e 0,1 ml da solução de verde de bromocresol SI. São necessários não mais que 0,5 ml da solução de hidróxido de sódio 0,1 M SV para promover viragem do indicador.
Cinzas sulfatadas (V.2.10). Determinar em 1 g da amostra. No máximo 0,1%.
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de ácido fórmico anidro, acetato de etila e cloreto de metileno (2:10:88), como fase móvel. Aplicar, separadamente,à placa, 5 μl de cada uma das soluções, recentemente preparadas, descritas a seguir.
Solução (1): dissolver 0,1 g da amostra em metanol e diluir a 5 ml com o mesmo solvente.
Solução (2): diluir l ml da solução (1) para 100 ml com metanol.
Desenvolver o cromatograma. Remover a placa, deixar secar sob corrente de ar quente e examinar sob luz ultravioleta (254 nm).
Qualquer mancha secundária observada no cromatograma da solução (1) não é mais intensa do que a mancha obtida no cromatograma da solução (2).
DOSEAMENTO
Transferir 2 g da amostra para um erlenmeyer provido de rolha esmerilhada, adicionar 40 ml de solução de hidróxido de sódio M.
AdapTar condensador de refluxo e aquecer cuidadosamente por 30 minutos. Esfriar. Lavar o condensador com 5 ml de água. Titular o excesso de hidróxido de sódio com solução de ácido sulfúrico 0,5 M determinando o ponto final potenciometricamente. Realizar ensaio em branco e fazer as correções necessárias. Cada ml de hidróxido de sódio M equivale a 152,1 mg de C10H12O3.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CATEGORIA
Conservante.
163
NORFLOXACINO
Norfloxacinum
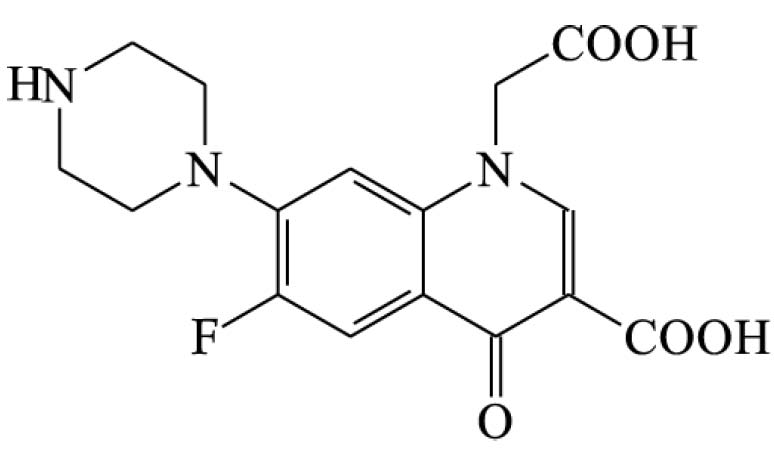
C16H18FN3O3 319,34 1344.01-3
Ácido 1-etil-6-fluor-1,4-diidro-4-oxo-7-(1-piperazinil)-3-quinolino carboxílico
Contém, no mínimo, 99,0% e, no máximo, 101,0% de C16H18FN3O3, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó cristalino branco a amarelo claro.
Solubilidade. Pouco solúvel em água, metanol, etanol, acetato de etila e acetona, facilmente solúvel em ácido acético, ligeiramente solúvel em clorofórmio e insolúvel em éter etílico.
Constantes físico-químicas
Faixa de fusão (V.2.2): entre 227 °C e 228 °C.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) da amostra dessecada até peso constante e dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de norfloxacino padrão, preparado de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14-3), na faixa de 200 a 400 nm, de uma solução de 0,0005% (p/V) em hidróxido de sódio 0,1 M, exibe máximo em 273 nm, idêntico ao observado no espectro de solução similar de padrão.
ENSAIOS DE PUREZA
Metais pesados (V.3.2.3 - Método II). No máximo 0,0015% (15 ppm).
Perda por dessecação (V.2.9). Secar sob pressão reduzida, que não exceda 5 mm de Hg a 100 °C, até peso constante. No máximo 1%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,1%.
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17) pelo método de difusão em ágar, utilizando cilindros.
Microrganismo: Staphylococcus epidermidis ATCC 12228.
Meios de cultura: meio de cultura número 1, para manutenção do microrganismo; solução salina estéril, para padronização do inóculo; e
meio de cultura número 11, para a camada base e preparação do inóculo.
Solução amostra: pesar, exatamente, cerca de 0,25 g de norfloxacino amostra e transferir quantitativamente para balão volumétrico de 250 ml com auxílio de 100 ml de hidróxido de sódio 0,1 M. Agitar por 15 minutos e completar o volume com tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2). Diluir, sucessivamente, até as concentrações de 10 μg/ml, 20 μg/ml e 40 μg/ml, utilizando água como diluente.
Solução padrão: pesar, exatamente, cerca de 50 mg de norfloxacino e transferir quantitativamente para balão volumétrico de 50 ml com auxílio de 20 ml de hidróxido de sódio 0,1 M. Agitar por 15 minutos e completar o volume com tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2). Diluir, sucessivamente, até as concentrações de 10 μg/ml, 20 μg/ml e 40 μg/ml, utilizando água como diluente.
Procedimento: adicionar 20 ml de meio número 11 em cada placa, esperar solidificar e adicionar 5 ml de inóculo a 0,7% e proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17.1), adicionando nos cilindros, 0,2 ml das soluções recentemente preparadas.
DOSEAMENTO
Proceder conforme descrito em titulação em meio não-aquoso
(V.3.4.5). Dissolver 0,46 g da amostra em 100 ml de ácido acético
glacial. Titular com ácido perclórico 0,1 M SV, determinando o ponto
final potenciometricamente. Realizar ensaio em branco e fazer as
correções necessárias. Cada ml de ácido perclórico 0,1 M SV equivale
a 31,93 mg de C16H18FN3O3.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz e umidade.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antimicrobiano.
163.1
NORFLOXACINO COMPRIMIDOS
Contém, no mínimo, 90,0% e, no máximo, 110,0%, da quantidade declarada de C16H18FN3O3.
IDENTIFICAÇÃO
A. Pesar e pulverizar os comprimidos. Proceder conforme descrito em Cromatografia de camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e mistura de clorofórmio-metanol-tolueno-dietilamina-água (40:40:20:14:8), como fase móvel. Aplicar, separadamente,à placa, 50 μl de cada uma das soluções, recentemente preparadas, descritas a seguir.
Solução (1): solução a 0,15% (p/V) de amostra em mistura de 1 parte de cloreto de metileno e 1 parte de metanol acidificado (ácido clorídrico 0,9% (V/V) em metanol). Centrifugar uma porção obtida e utilizar o sobrenadante límpido.
Solução (2): solução a 0,15% (p/V) de norfloxacino padrão preparada no mesmo solvente da solução (1). Centrifugar uma porção obtida e utilizar o sobrenadante límpido.
Procedimento: desenvolver o cromatograma. Remover a placa, deixar secar ao ar e examinar sob luz ultravioleta (254 nm). A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (2).
B. Proceder conforme descrito no método B de Doseamento. O tempo de retenção do pico principal obtido com a solução amostra correspondeàquele do pico principal da solução padrão.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: tampão pH 4,0, 750 ml
Aparelhagem: pás, 50 rpm
Tempo: 30 minutos
Procedimento: após o teste, retirar alíquota do meio de dissolução, filtrar e diluir em tampão pH 4,0 até concentração adequada. Medir as absorvâncias em 278 nm (V.2.14.-3), utilizando tampão pH 4,0 para ajuste do zero. Calcular a quantidade de C16H18FN3O3 dissolvido no meio, comparando as leituras obtidas com a da solução padrão na concentração de 0,005% (p/V), preparada no mesmo solvente.Tolerância: não menos do que 80% (T) da quantidade declarada de C16H18FN3O3 se dissolvem em 30 minutos.
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito em Determinação da potência na monografia de norfloxacino. Preparar a solução amostra como descrito a seguir.
Solução amostra: pesar e pulverizar 20 comprimidos. Utilizar quantidade de pó equivalente a 0,25 g de norfloxacino e transferir quantitativamente para balão volumétrico de 500 ml com auxílio de 200 ml de hidróxido de sódio 0,1 M. Agitar por 15 minutos, completar o volume com tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) e filtrar. Diluir, sucessivamente, até concentrações de 10μg/ml, 20 μg/ml e 40 μg/ml, utilizando água como diluente.
DOSEAMENTO
A. Por espectrofotometria de absorção no ultravioleta. Pesar e pulverizar 20 comprimidos. Utilizar quantidade do pó equivalente a 50 mg de norfloxacino e transferir para balão volumétrico de 100 ml com o auxílio de 50 ml de ácido clorídrico 0,1 M. Agitar por 15 minutos, completar o volume com o mesmo diluente e filtrar. Diluir, sucessivamente, até a concentração de 0,0005% (p/V), utilizando ácido clorídrico 0,1 M como solvente. Preparar solução padrão na mesma concentração, utilizando o mesmo solvente. Medir as absorvâncias das soluções em 277 nm (V.2.14.-3), utilizando ácido clorídrico 0,1 M para o ajuste do zero. Calcular a quantidade de C16H18FN3O3 nos comprimidos, a partir das leituras obtidas.
B. Por cromatografia líquida de alta eficiência. Proceder conforme
descrito em Cromatografia líquida de alta eficiência (V.2.17.4), utilizando
cromatógrafo provido de detector ultravioleta a 275 nm, coluna
de 300 mm de comprimento e 3,9 mm de diâmetro interno,
empacotada com sílica quimicamente ligada a octadecilsilano (5 μm
ou 10 μm), mantida a 40 °C; fluxo da fase móvel de 2,0 ml/minuto.
Fase móvel: mistura de ácido fosfórico 0,1% (V/V) e acetonitrila
(85:15). Anteriormente à análise, estabilizar a coluna com solução de
fosfato de sódio monobásico 0,01 M ajustado com ácido fosfórico
para pH 4,0 a um fluxo de 0,5 ml/minuto por 8 horas.
Solução amostra: pesar e pulverizar 20 comprimidos. Utilizar quantidade
de pó equivalente a 0,1 g de norfloxacino. Transferir para
balão volumétrico de 200 ml com auxílio de 80 ml de fase móvel,
submeter ao ultra-som por 10 minutos, completar o volume comácido fosfórico 0,1% (V/V) e filtrar. Diluir, sucessivamente, até concentração
de 20 μg/ml, utilizando fase móvel como solvente.
Solução padrão: pesar, exatamente, cerca de 50 mg de norfloxacino.
Transferir para balão volumétrico de 50 ml com auxílio de 20 ml de fase móvel, agitar por 15 minutos, completar o volume com o mesmo solvente. Diluir, sucessivamente, até concentração de 20 μg/ml, utilizando fase móvel como solvente.
A eficiência da coluna não deve ser menor que 5 000 pratos téoricos/metro. O fator de cauda não deve ser superior a 2. O desvio
padrão relativo das áreas de replicatas dos picos registrados não deve
ser maior que 2,0%.
Procedimento: injetar separadamente 10 μl das soluções amostra e padrão, registrar os cromatogramas e medir as áreas dos picos. Calcular a quantidade de C16H18FN3O3 na solução amostra a partir das respostas obtidas com as soluções padrão e amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados e protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
_________________________________________________
XII.4. TAMPÕES
Tampão pH 4,0
Preparação - Transferir 900 ml de água para um balão volumétrico de 1 000 ml, adicionar 2,86 ml de ácido acético glacial e 1 ml de uma solução de hidróxido de sódio 50% (p/p), completar o volume comágua e homogeneizar. Se necessário, ajustar o pH com ácido acético glacial ou solução de hidróxido de sódio 50% (p/p).
164
NOZ-DE-COLA
Semen colae
Cola nitida (Vent.) A.Chev. - STERCULIACEAE
A droga vegetal consiste dos cotilédones dessecados de Cola nitida (Vent.) A. Chev. contendo, no mínimo, 1,7% de taninos totais e 2,0% de cafeína.
SINONÍMIA CIENTÍFICA
Sterculia nitida Vent. e Cola vera K.Schum.
SINONÍMIA VULGAR
Cola, semente-de-cola.
CARACTERES ORGANOLÉPTICOSOs cotilédones apresentam sabor adstringente e algo amargo e odor quase nulo.
DESCRIÇÃO MACROSCÓPICA
Os cotilédones são em número de dois, normalmente encontrados no comércio já separados. São duros e desiguais, sólidos, irregulares, de cor castanho-avermelhada, de tamanho muito variável, com 2 cm a 5 cm de comprimento por cerca de 2 cm de largura e até 1 cm de espessura. O ápice do cotilédone é mais largo do que a sua base e ambos são arredondados. A margem é inteira. A superfície externa de cada cotilédone é convexa ou ligeiramente deprimida, rugosa, de coloração castanha a castanho-avermelhada. A superfície interna é plana ou deprimida, mais ou menos lisa, geralmente irregular, apresentando na base pequena cavidade, contendo, às vezes, a radícula e a plúmula, ou vestígios destas. A superfície de fratura é uniforme e castanha brilhante.
DESCRIÇÃO MICROSCÓPICA
Os cotilédones estão envoltos por uma epiderme formada por células retangulares, pequenas ou ligeiramente alongadas no sentido radial e são constituídos por um parênquima homogêneo de células poligonais,às vezes de contorno irregular. As células mais internas são maiores, com paredes espessas e pontoadas, de coloração castanha, contendo compostos fenólicos, matéria graxa e abundantes grãos de amido. Esses últimos, estão principalmente distribuídos nas células centrais e são desiguais, esféricos, ovais, ovais-arredondados, oblongos, reniformes, elipsóides ou piriformes, com hilo ramificado, centralizado ou excêntrico, quase sempre fundido, em forma de estrela ou de cruz e suas estrias concêntricas são pouco visíveis. O tamanho dos grãos varia de 5 μm a 35 μm, raramente 45 μm.
DESCRIÇÃO MICROSCÓPICA DO PÓ
O pó atende a todas as exigências estabelecidas para a espécie, menos os caracteres macroscópicos. São característicos: cor castanho-avermelhada a moderadamente amarelado-acastanhada; fragmentos de epiderme e de parênquima com células poligonais, de paredes pardas ou castanho-avermelhadas, contendo numerosos e variados grãos de amido, como os descritos; escassos fragmentos de pequenos feixes fibrovasculares. Os grãos de amido, quando observados em luz polarizada, exibem uma cruz na região do hilo.
IDENTIFICAÇÃO
A. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando cromatoplaca de sílica-gel GF254 e acetato de etila-metanol-água (100:13,5:10), como fase móvel. Aplicar, separadamente, em forma de banda, 5-10 μl da solução amostra e 2-3 μl da solução de referência, preparadas como descrito a seguir.
Solução amostra: extrair a droga previamente pulverizada, sob refluxo durante 15 minutos, em concentração igual a 2% (p/V), usando etanol como líquido extrator. Filtrar e aplicar na cromatoplaca.
Solução referência: dissolver 10 mg de cafeína em 2 ml de etanol absoluto.
Desenvolver o cromatograma em percurso de 15 cm. Remover a cromatoplaca e deixar secar ao ar por alguns minutos. Observar sob luz ultravioleta (254 nm). A mancha com Rf próximo a 0,50, corresponde a cafeína. Nebulizar com o reagente de Dragendorff SR.
Adicionalmente nebulizar com solução aquosa de nitrito de sódio a 5% (p/V). A mancha correspondente a cafeína apresenta coloração vermelho tijolo.
ENSAIOS DE PUREZA
Matéria estranha (V.4.2.2.). No máximo 3%.
Determinação de água (V.4.2.3.). No máximo 15%.
Cinzas totais (V.4.2.4.). No máximo 5%.
DOSEAMENTO
A. Determinar o teor de cafeína. Pesar exatamente cerca de 0,25 g da amostra pulverizada. Extrair com 20 ml de solução de ácido sulfúrico 2,5% sob agitação mecânica durante 15 minutos, por quatro vezes.
Filtrar as porções para balão volumétrico de 100 ml. Completar o volume com solução de ácido sulfúrico 2,5% (V/V) e transferir 10 ml desta solução para balão volumétrico de 100 ml. Completar o volume com a mesma solução de ácido sulfúrico, obtendo-se, assim, concentração teórica em torno de 15 μg/ml.
Solução de referência: pesar 500 μg de cafeína e dissolver em 100 ml de solução de ácido sulfúrico 2,5 % (V/V). Empregar 3 ml desta solução, equivalentes a 1,5 mg de cafeína, completar o volume em balão volumétrico de 100 ml com solução de ácido sulfúrico 2,5% (V/V), obtendo-se a concentração de 15,0 μg/ml de cafeína.
Medida da absorvância: empregar cubetas de 1 cm. Efetuar a leitura em espectrofotômetro a 271 nm. Utilizar solução de ácido sulfúrico 2,5% (V/V), como solução de compensação. Calcular o teor de cafeína (metil-xantinas) utilizando a equação:
Em que
C = Teor de rendimento de metilxantinas na amostra;
AA = Absorvância da solução amostra;
AP = Absorvância da solução referência;
CP = Concentração da solução referência em μg/ml;
MA = Massa em gramas de amostra;
10 = Fator de diluição.
B. Determinar o teor de taninos totais: proteger da luz as amostras durante a extração e a diluição. Utilizar água isenta de dióxido de carbono em todas as operações. Pesar 0,75 g da droga pulverizada, transferir para erlenmeyer e adicionar 150 ml de água. Aquecer até fervura e manter em banho-maria à temperatura de 80-90 oC por 30 minutos. Resfriar em água corrente, transferir a mistura para balão volumétrico e diluir a 250 ml com água. Deixar decantar o sedimento e filtrar através de papel filtro. Desprezar os primeiros 50 ml do filtrado.
Polifenóis totais: diluir 5 ml do filtrado para 25 ml com água. Misturar 5 ml desta solução com 2 ml da solução de ácido fosfotúngstico SR e diluir a 50 ml com solução de carbonato de sódio 10,6% (p/V).
Medir a absorvância da solução (A1) a 715 nm (V.2.14), exatamente 3 minutos após a adição do último reagente, utilizando água como branco.
Polifenóis não adsorvidos pelo pó-de-pele: adicionar a 20 ml do filtrado 0,2 g de pó-de-pele e agitar vigorosamente por 60 minutos.
Filtrar. Diluir 5 ml do filtrado a 25 ml com água. Misturar 5 ml desta solução com 2 ml da solução de ácido fosfotúngstico SR e diluir 50 ml com solução de carbonato de sódio 10,6% (p/V). Medir a absorvância da solução a 715 nm (A2) (V.2.14), exatamente 3 minutos após a adição do último reagente, utilizando água como branco.
Solução referência: dissolver 50 mg de pirogalol em água e diluir a 100 ml. Diluir 5 ml desta solução a 100 ml com água. Misturar 5 ml desta solução com 2 ml da solução de ácido fosfotúngstico SR e diluir a 50 ml com solução de carbonato de sódio 10,6% (p/V). Medir a absorvância desta solução a 715 nm (A3) (V.2.14), exatamente 3 minutos após a adição do último reagente e dentro de 15 minutos contados da dissolução do pirogalol, utilizando água como branco.
Calcular o teor de taninos pela expressão:
Em que
m = massa da amostra em gramas.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, ao abrigo da luz e do calor.
________________________________________________________
XII.2 REAGENTE E SOLUÇÕES REAGENTES
Ácido fosfotúngstico SR
Aquecer 10 g de tungstato de sódio sob refluxo por 3 horas com 8 ml de ácido fosfórico 85% (V/V) e 75 ml de água. Após resfriamento, diluir com água para 100 ml.
LEGENDA
Figura 1: Cola nitida (Vent.) A.Chev. - A. aspecto da face externa do cotilédone; B. aspecto da face interna do cotilédone; C. cotilédone em vista equatorial; D, E e F. detalhe de células parenquimáticas, encontradas no pó, evidenciando tamanhos variáveis de células e paredes pontoadas; G. detalhe de grãos de amido, mostrando variabilidade quanto a forma, tamanho e hilo. As escalas correspondem em A, B, C a 2 cm; em D, E e F a 100 μm; em G a 50 μm.
165
OFLOXACINO
Ofloxacinum
Ácido (±)-9-fluor-2,3-diidro-3-metil-10-(4-metil-1-piperazinil)-7-oxo- 7H pirido[1,2,3-de]-1,4- benzoxazina -6-carboxílico.
Contém, no mínimo, 98,5% e, no máximo, 101,5% de C18H20FN3O4, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Cristais em forma de agulhas incolores.
Constantes físico-químicas
Faixa de fusão (V.2.2 ): 250 °C a 257 °C, com decomposição.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) da amostra dessecada até peso constante e dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de ofloxacino padrão, preparado de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 200 a 400 nm, de uma solução a 0,00067% (p/V) em ácido clorídrico 0,1 M exibe máximos idênticos aos observados no espectro de solução similar de ofloxacino padrão.
ENSAIOS DE PUREZA
Poder rotatório (V.2.8). De +1° a -1°. Determinar em solução a 1% (p/V) em clorofórmio, em relação à substância dessecada.
Metais pesados (V.3.2.3-Método II). No máximo 0,001% (10 ppm).
Perda por dessecação (V.2.9). Determinar em 2 g de amostra, em estufa a 105 ºC, por 4 horas. No máximo 0,2%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,1%.
Arsênio (V.3.2.5 - Método II). No máximo 0,0001% (1 ppm).
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17) pelo método difusão em ágar, utilizando cilindros.
Microrganismo: Micrococcus luteus ATCC 9341.
Meios de cultura: meio número 1, para manutenção do microrganismo;
solução salina estéril, para a padronização do inóculo e meio número 11, para a camada base e camada de inóculo na placa.
Solução amostra: pesar, exatamente, cerca de 0,25 g de amostra, transferir para balão volumétrico de 250 ml com auxílio de 100 ml de tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), agitar por 30 minutos e completar o volume com o mesmo diluente. Diluir, sucessivamente, até as concentrações de 20 μg /ml, 30 μg/ml e 45 μg /ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), como diluente.
Solução padrão: pesar, exatamente, cerca de 50 mg de ofloxacino padrão, transferir para balão volumétrico de 50 ml e completar o volume com tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2). Diluir, sucessivamente, até as concentrações de 20 μg /ml, 30 μg /ml e 45 μg /ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), como diluente.
Procedimento: adicionar 20 ml de meio de cultura número 11 em cada placa, esperar solidificar, adicionar 5 ml de inóculo a 0,5% e proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17), adicionando aos cilindros 0,2 ml das soluções recentemente preparadas.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antimicrobiano.
165.1
OFLOXACINO COMPRIMIDOS
Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de C18H20FN3O4.
IDENTIFICAÇÃO
Pesar e pulverizar os comprimidos. Preparar solução a 0,00067% (p/V) em ácido clorídrico 0,1 M, agitar mecanicamente por 20 minutos e filtrar. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 200 a 400 nm, exibe máximos idênticos aos observados no espectro de solução similar de ofloxacino padrão.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito em Determinação da potência na monografia de ofloxacino. Preparar a solução amostra como descrito a seguir.
Solução amostra: pesar e pulverizar 20 comprimidos. Utilizar quantidade de pó equivalente a de 0,25 g de ofloxacino, transferir quantitativamente para balão volumétrico de 250 ml com auxílio de 100 ml de tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2).
Agitar por 30 minutos e completar o volume com o mesmo diluente e filtrar. Diluir, sucessivamente, até as concentrações de 20 μg/ml, 30μg /ml e 45 μg /ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), como diluente.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
165.2
OFLOXACINO SOLUÇÃO INJETÁVEL
Contém, no mínimo 90,0% e, no máximo,110,0% da quantidade declarada de C18H20FN3O4. Pode ser preparada em água para injetáveis ou em outro solvente adequado.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14-4) da amostra previamente levada a resíduo seco em rotavapor, sob pressão reduzida, mantida em dessecador e dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de ofloxacino padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel 60 GF254, como suporte, e mistura de cloreto de metileno-metanol-hidróxido de amônio-acetonitrila (40:40:20:10), como fase móvel. Aplicar, separadamente, à placa, 5μl de cada uma das soluções recentemente preparadas, descritas a seguir.
Solução (1):solução a 0,05% (p/V) da amostra em água.
Solução (2): solução a 0,05% (p/V) de ofloxacino padrão em água.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e observar sob luz ultravioleta (254 nm e 336 nm). A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidadeàquela obtida com a solução (2).
CARACTERÍSTICAS
Determinação de volume (V.1.2). Cumpre o teste.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1). Cumpre o teste.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 10 ml/kg de uma solução contendo 2 mg/ml de ofloxacino.
Endotoxinas bacterianas (V.5.1.9). Cumpre o teste. No máximo 5 UE/mg de ofloxacino.
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito em Determinação da potência na monografia de ofloxacino. Preparar a solução amostra como descrito a seguir.
Solução amostra: transferir volume da solução injetável equivalente a 0,25 g de ofloxacino para balão volumétrico de 250 ml e adicionar 100 ml de tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2). Agitar por 30 minutos e completar o volume com o mesmo diluente. Diluir, sucessivamente, até as concentrações de 20 μg/ml, 30μg /ml e 45 μg /ml, utilizando tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), como diluente.
EMBALAGEM E ARMAZENAMENTO
Em recipientes de vidro tipo I, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
166
OXAMNIQUINA
Oxamniquinum
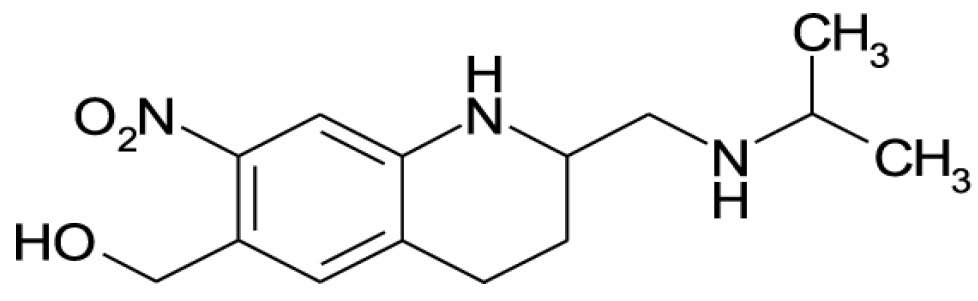
C14H21N3O3 279,34 21738-42-1
(±)1,2,3,4-tetraidro-2-[[(1-metiletil)-amino]metil]-7-nitro-6-quinolinometanol
Contém, no mínimo, 97,0% e, no máximo, 103,0% de C14H21N3O3, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó muito fino, de cor amarelada, inodoro.
Solubilidade. Praticamente insolúvel em água, solúvel em etanol e metanol, pouco solúvel em clorofórmio e acetona. Facilmente solúvel em ácido acético, praticamente insolúvel em solução de hidróxido de sódio.
Constantes físico-químicas
Faixa de fusão (V.2.2): 146 ºC a 151 ºC, com decomposição.
IDENTIFICAÇÃO
Os testes de identificação A e B podem ser omitidos se forem realizados os testes C, D, E, F e G. Os testes de identificação C, D, E, F e G podem ser omitidos se forem realizados os testes A e B.
A. O espectro de absorção no infravermelho (V.2.14.-4) da amostra dispersa em brometo de potássio apresenta máximos de absorção nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de oxamniquina padrão, preparado de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 220 a 400 nm, de solução da amostra a 0,001% (p/V), exibe máximo de absorção em 251 nm + 1 nm.
C. Dissolver 1 g de dicromato de potássio em solução previamente preparada de ácido sulfúrico em água (1:3). Dissolver 15 mg da amostra em 20 gotas de acetona. Adicionar 5 a 8 gotas da solução de dicromato de potássio e agitar. Forma-se precipitado verde, em 5 segundos.
D. Colocar em vidro de relógio, cerca de 10 mg da amostra. Adicionar 2 gotas de formaldeído 37% (V/V) e 10 gotas de ácido sulfúrico.
Desenvolve-se coloração verde.
E. Misturar, em tubo de ensaio, 10 mg da amostra e 1,5 ml de solução recém-preparada de sulfato ferroso amoniacal 5% (p/V). Adicionar 1 gota de ácido sulfúrico 3 M e 1 ml de solução metanólica de hidróxido de potássio 2 M. Tampar o tubo, agitar bem e observar.
Ocorre, rapidamente, mudança de cor de azul para marrom.
F. Em tubo de ensaio, dissolver cerca de 10 mg da amostra em ácido clorídrico M, acrescentar óxido de magnésio. Aquecer, se necessário.
Desprendem-se, aos poucos, vapores alcalinos que escurecem o papel de prata-manganês colocado na extremidade superior do tubo.
G. Proceder conforme descrito em Cromatografia em camada delgada
(V.2.17.1), utilizando placas de sílica-gel GF254, como suporte e mistura de clorofórmio, n-hexano-álcool isopropílico- hidróxido de amônio
(100:50:10:7,5), como fase móvel. Aplicar, separadamente, sobre a placa, 20 μl de cada uma das soluções descritas a seguir.
Solução (1): dissolver 25 mg da amostra em clorofórmio e completar o volume para 10 ml. Transferir 1 ml da solução anterior para balão volumétrico de 25 ml e completar o volume com clorofórmio.
Solução (2): dissolver 25 mg de oxamniquina padrão em clorofórmio e completar o volume para 10 ml. Desta solução, transferir 1 ml para balão volumétrico de 25 ml e completar o volume com clorofórmio.
Desenvolver o cromatograma por 10 cm. Remover a placa, deixar
secar ao ar e observar sob luz ultravioleta (254 nm). A mancha
principal obtida com a solução (1) corresponde em posição e intensidadeàquela obtida com a solução( 2).
ENSAIOS DE PUREZA
Poder rotatório (V.2.8). Entre -4º e +4º, a 25 ºC. Determinar em solução a 2% (p/V) em tetraidrofurano.
pH (V.2.19). 8,0 a 10,0. Determinar na suspensão aquosa a 1% (p/V).
Perda por dessecação (V.2.9). Determinar em 1 g de amostra, em estufa a 105 ºC por 2 horas. No máximo 1%.
Cinzas sulfatadas (V.2.10). Determinar em 2 g da amostra. Incinerar a 600 ºC por 2 horas. No máximo 0,2%.
Ferro (V.3.2.4). Dissolver o resíduo obtido em Cinzas sulfatadas em 4 ml de ácido clorídrico, cuidadosamente, com ligeiro aquecimento.
Diluir com água para 100 ml. Utilizar 10 ml desta solução e prosseguir
conforme descrito em Ensaio-limite para ferro. No máximo
0,005% (50 ppm).
Metais pesados (V.3.2.3 - Método II). No máximo 0,005% (50 ppm).
DOSEAMENTO
A. Por titulação em meio não-aquoso (V.3.4.5). Pesar, exatamente, cerca de 0,1 g de amostra e dissolver em 50 ml de ácido acético glacial e 5 ml de anidrido acético. Acrescentar gotas de vermelho de quinaldina SI e titular com ácido perclórico 0,1 M SV até viragem de vermelho para incolor. Realizar ensaio em branco e proceder à correções necessárias. Cada ml de ácido perclórico 0,1 M SV equivale a 27,934 mg de C14H21N3O3.
B. Por espectrofotometria de absorção no ultravioleta (V.2.14-3). Pesar, exatamente, cerca de 20 mg da amostra e dissolver em metanol para 100 ml. Diluir, sucessivamente, em metanol de modo a obter concentração final de 0,001% (p/V). Preparar solução padrão na mesma concentração, utilizando o mesmo solvente. Medir as absorvâncias das soluções resultantes em 251 nm, utilizando metanol como branco.
Calcular o teor de C14H21N3O3 na amostra a partir das leituras obtidas.
CLASSE TERAPÊUTICA
Esquistossomicida.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, ao abrigo da luz.
ROTULAGEM
Observar a legislação vigente.
167
PARACETA MOL
Paracetamolum
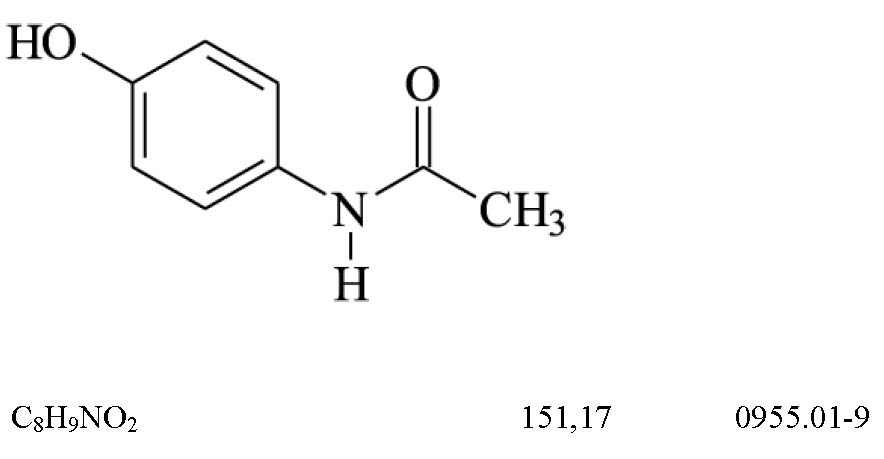
N-(4-hidroxifenil)acetamida
Contém, no mínimo, 98,0% e, no máximo, 101,0% de C8H9NO2 , em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Pó cristalino, branco, inodoro, com leve sabor amargo.
Solubilidade. Ligeiramente solúvel em água, solúvel em água fervente, facilmente solúvel em etanol, praticamente insolúvel em clorofórmio e éter etílico. Solúvel em hidróxido de sódio M.
Constantes físico-químicas
Faixa de fusão (V.2.2): 168 ºC a 172 ºC.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4) da amostra dessecada sobre sílica-gel, e dispersa em brometo de potássio apresenta máximos de absorção nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de paracetamol padrão, preparado de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14.-3), na faixa de 200 a 400 nm, da solução da amostra a 0,0005% (p/V) em mistura deácido clorídrico 0,1 M e metanol (1:100), exibe máximos e mínimos somente nos mesmos comprimentos de onda de solução similar de paracetamol padrão.
C. A 10 ml de uma solução a 1% (p/V) da amostra adicionar uma gota de cloreto férrico SR. Deve desenvolver-se cor azul-violácea.
ENSAIOS DE PUREZA
pH (V.2.19). 5,3 a 6,5. Determinar na solução saturada.
Água (V.2.20.1). No máximo 0,5%.
Resíduo por incineração (V.2.10). No máximo 0,1%.
Cloreto (V.3.2.1). Agitar 1 g da amostra com 25 ml de água, filtrar e adicionar 1 ml de ácido nítrico 2 M. Prosseguir conforme descrito em Ensaio-limite para cloretos. No máximo 0,014% (140 ppm).
Sulfato (V.3.2.2). Agitar 1 g da amostra com 25 ml de água, filtrar quantitativamente para tubo de Nessler e proceder conforme descrito em Ensaio-limite para sulfatos. No máximo 0,02% (200 ppm).
Sulfeto. Pesar cerca de 2,5 g da amostra em béquer de 50 ml. Adicionar 5 ml de etanol e 1 ml de ácido clorídrico M. Umedecer comágua um papel de filtro impregnado com acetato de chumbo e colocar sobre vidro de relógio. Cobrir o béquer com o vidro de relógio de tal forma que uma das pontas do papel fique na abertura do frasco.
Aquecer em chapa elétrica até ebulição. Nenhuma mancha ou coloração aparece no papel com acetato de chumbo.
Metais pesados (V.3.2.3.-3 - Método II). Dissolver 1 g da amostra em mistura de 85 partes de acetona e 15 partes de água. Completar para20 ml usando a mesma mistura de solventes. Transferir 12 ml da solução obtida para tubo de Nessler e proceder conforme descrito emEnsaio-limite para metais pesados (V.3.2.3.-3). No máximo 0,002% (20 ppm).
Substâncias facilmente carbonizáveis. Dissolver 0,5 g da amostra em 5 ml de ácido sulfúrico. A cor da solução (V.2.12) não é mais intensa que a da solução padrão de cor A.
Aminofenol livre. Dissolver 0,5 g da amostra numa mistura de metanol-água (1:1) e completar o volume para 10 ml com a mesma
mistura de solventes. Preparar 10 ml de solução padrão contendo 0,5
g de paracetamol padrão isento de 4-aminofenol e 0,5 ml de solução
de 4-aminofenol a 0,005% (p/V), na mesma mistura de solventes.
Adicionar, simultaneamente, à solução amostra e à solução padrão, 0,2 ml de solução de carbonato de sódio anidro a 1% (p/V), recentemente preparada. Homogeneizar e deixar em repouso durante 30 minutos. A solução problema não é mais corada de azul que a solução padrão.
Cloroacetanilida. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1). Preparar a fase estacionária dissolvendo 8 mg de acetato de sódio em 50 ml de água, adicionando, em seguida, 20 g de sílica-gel GF254. Preparar placas com 0,5 mm de espessura. Utilizar como fase móvel mistura de clorofórmio-benzeno-acetona (65:10:25). Aplicar separadamente à placa, 100 μl da Solução (1) e 20 μl da Solução (2), descritas a seguir.
Solução (1): transferir 1 g da amostra para um tubo de centrífuga de 15 ml com tampa, adicionar 5 ml de éter etílico, agitar mecanicamente por 30 minutos e centrifugar a 1 000 rpm por 15 minutos ou até obter separação nítida.
Solução (2): solução de p-cloroacetanilida a 10 μg/ml em etanol.
Desenvolver o cromatograma em sistema aberto até a fase móvel atingir pelo menos 12 cm da origem. Remover a placa, deixar secar ao ar e manter, por 30 minutos, sob luz ultravioleta (254 nm) a uma distância de 4 cm. Localizar as manchas sob luz ultravioleta de 365 nm. Qualquer mancha fluorescente azul produzida pela Solução (1), com Rf entre 0,5 e 0,6 não é maior ou mais intensa que aquela produzida pela Solução (2). No máximo 0,001%.
DOSEAMENTO
Pesar, exatamente, cerca de 0,15 g da amostra, dissolver em 50 ml de hidróxido de sódio 0,1 M, adicionar 100 ml de água, agitar mecanicamente por 15 minutos e adicionar água suficiente para 200 ml.
Homogeneizar, filtrar e diluir 10 ml do filtrado para 100 ml comágua. Transferir 10 ml da solução resultante para balão volumétrico
de 100 ml, adicionar 10 ml de hidróxido de sódio 0,1 M e completar
o volume com água. Preparar solução padrão de paracetamol em
hidróxido de sódio 0,01 M, na mesma concentração final. Medir as
absorvâncias das soluções resultantes em 257 nm (V.2.14.-3), utilizando
hidróxido de sódio 0,01 M para ajuste do zero. Calcular o
teor de C8H9NO2 na amostra a partir das leituras obtidas. Alternativamente,
realizar os cálculos considerando A(1%,1cm) = 715, em
257 nm.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados e opacos.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Analgésico e antipirético.
167.1
PARACETAMOL COMPRIMIDOS
Contém, no mínimo, 95,0% e, no máximo, 105,0% da quantidade declarada de C8H9NO2.
IDENTIFICAÇÃO
A. Pesar e pulverizar os comprimidos. Extrair quantidade do pó equivalente a 0,5 g de paracetamol com 20 ml de acetona. Filtrar, evaporar o filtrado e secar a 105 oC. O espectro de absorção no infravermelho (V.2.14.-4) do resíduo, disperso em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles obtidos no espectro de paracetamol padrão, preparado de maneira idêntica.
B. Aquecer até ebulição 0,1 g do resíduo obtido no teste A de Identificação com 1 ml de ácido clorídrico por três minutos, adicionar 10 ml de água e resfriar. Nenhum precipitado é produzido. Adicionar 0,05 ml de dicromato de potássio 0,0167 M. Desenvolve-se coloração violácea, que não muda para vermelha.
C. O ponto de fusão do resíduo obtido no teste A de Identificação é de, aproximadamente, 169 oC.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: tampão fosfato pH 5,8, 900 ml
Aparelhagem: cesta, 50 rpm
Tempo: 30 minutos
Procedimento: após o teste, retirar alíquota do meio de dissolução, filtrar e diluir em tampão fosfato pH 5,8 até concentração adequada.
Medir as absorvâncias das soluções resultantes em 243 nm (V.2.14.- 3), em comparação com uma solução de paracetamol padrão a 0,0017% (p/V) em tampão fosfato pH 5,8. Utilizar o mesmo solvente para o ajuste do zero. Calcular a quantidade de C8H9NO2 dissolvido no meio a partir das leituras obtidas.
Tolerância: não menos que 80% (T) da quantidade declarada de C8H9NO2 se dissolvem em 30 minutos.
ENSAIOS DE PUREZA
p-Aminofenol. Proceder conforme descrito em Cromatografia líquida de alta eficiência (V.2.17.4), utilizando cromatógrafo provido de detector ultravioleta a 272 nm; coluna de 200 mm de comprimento e 4,6 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octadecilsilano (10 μm); fluxo da fase móvel de 2 ml/minuto.
Fase móvel: preparar solução de butanossulfonato de sódio 0,1 M utilizando como solvente mistura água-metanol-ácido fórmico (85:15:0,4).
Solução (1): pesar e pulverizar os comprimidos. Transferir quantidade do pó equivalente a 1 g de paracetamol para balão volumétrico de 100 ml, adicionar 15 ml de metanol e agitar. Completar o volume com água, homogeneizar e filtrar.
Solução (2): preparar solução a 0,001% (p/V) de p-aminofenol em metanol 15% (V/V).
Procedimento: injetar, separadamente, 20 μl da solução (1) e da solução (2), registrar os cromatogramas e medir as áreas dos picos. A área do pico correspondente ao p-aminofenol obtido no cromatograma com a solução (1) não é maior que o pico principal obtido no cromatograma com a solução (2).
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada, utilizando sílica-gel GF254, como suporte e mistura de clorofórmio-acetona-tolueno (65:25:10) como fase móvel.
Aplicar, separadamente, à placa, 200 μl da solução (1) e 40 μl de cada uma das soluções (2), (3) e (4), descritas a seguir.
Solução (1): pesar e pulverizar os comprimidos. Transferir quantidade do pó equivalente a 1 g de paracetamol para um tubo de centrífuga de 15 ml com tampa de vidro esmerilhada. Adicionar 5 ml de éter etílico e agitar mecanicamente por 30 minutos. Centrifugar a 1 000 rotações por minuto, durante 15 minutos ou até obter sobrenadante límpido.
Utilizar o sobrenadante.
Solução (2): diluir 1 ml da solução (1) para 10 ml com etanol.
Solução (3): preparar solução a 0,005% (p/V) de p-cloroacetanilida em etanol.
Solução (4): dissolver 0,25 g de p-cloroacetanilida e 0,1 g de paracetamol em etanol suficiente para produzir 100 ml.
Desenvolver o cromatograma em cuba não saturada, até a fase móvel atingir 14 cm da origem. Remover a placa, secar com o auxílio de corrente de ar quente e examinar sob luz ultravioleta (254 nm).
Qualquer mancha correspondente à p-cloroacetanilida obtida no cromatograma com a solução (1) não é mais intensa que a mancha obtida no cromatograma com a solução (3). Qualquer mancha secundária obtida no cromatograma com a solução (2) com valor de Rf inferior ao da p-cloroacetanilida não é mais intensa que a mancha obtida no cromatograma com a solução (3). O teste só é válido se o cromatograma obtido com a solução (4) mostrar duas manchas principais nitidamente separadas, sendo que a mancha correspondente a p-cloroacetanilida apresenta Rf de maior valor.
DOSEAMENTO
Pesar e pulverizar 20 comprimidos. Tranferir quantidade do pó equivalente a 0,15 g de paracetamol para balão volumétrico de 200 ml.
Adicionar 50 ml de hidróxido de sódio 0,1 M, 100 ml de água, agitar mecanicamente por 15 minutos e completar o volume com água.
Homogeneizar, filtrar e diluir 10 ml do filtrado para 100 ml comágua. Transferir 10 ml da solução resultante para balão volumétrico
de 100 ml, adicionar 10 ml de hidróxido de sódio 0,1 M e completar
o volume com água. Preparar solução padrão de paracetamol em
hidróxido de sódio 0,01 M, na mesma concentração final. Medir as
absorvâncias das soluções resultantes em 257 nm (V.2.14.-3), utilizando
hidróxido de sódio 0,01 M para ajuste do zero. Calcular a
quantidade de C8H9NO2 nos comprimidos a partir das leituras obtidas.
Alternativamente, realizar os cálculos considerando A(1%,1cm) = 715, em 257 nm.
EMBALAGEM E ARMAZENAMENTO
Conservar em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
167.2
PARACETAMOL SOLUÇÃO ORAL
Contém, no mínimo, 90,0% e, no máximo, 110,0% de C8H9NO2.
IDENTIFICAÇÃO
A. Transferir volume da solução oral equivalente a 0,1 g de paracetamol
para balão volumétrico de 100 ml. Completar o volume
com metanol e homogeneizar. Transferir 1 ml da solução resultante
para balão volumétrico de 100 ml, adicionar 1 ml de ácido clorídrico
0,1 M, completar o volume com metanol e homogeneizar. Proteger a
solução da luz e, imediatamente, traçar o espectro de absorção na
faixa de 200 a 400 nm (V.2.14.-3). O espectro resultante apresenta
máximo de absorção em 249 nm.
B. Transferir para funil de separação volume da solução oral equivalente a 0,1 g de paracetamol. Adicionar 3 ml de ácido clorídrico 0,1 M, agitar, extrair com 10 ml de éter etílico e evaporar o extrato orgânico até cerca de 1 ml. Adicionar 2 ml de ácido clorídrico 0,1 M e continuar evaporação do éter etílico. Esfriar e adicionar 2 gotas de dicromato de potássio SR ao resíduo final. Desenvolve-se coloração laranja escura.
CARACTERÍSTICAS
Determinação de volume (V.1.2). Cumpre o teste.
pH (V.2.19). 3,8 a 6,1.
ENSAIOS DE PUREZA
p-Aminofenol. Proceder conforme descrito em p-aminofenol, na monografia de paracetamol comprimidos. Utilizar as soluções descritas a seguir.
Solução (1): agitar um volume da solução oral equivalente a 0,5 g de paracetamol com 15 ml da fase móvel e diluir para 25 ml com o mesmo solvente. Filtrar se necessário.
Solução (2): preparar solução a 0,0024% (p/V) de p-aminofenol na fase móvel.
DOSEAMENTO
A. Por espectrofotometria de absorção no ultravioleta. Transferir volume da solução oral equivalente a 0,1 g de paracetamol para balão volumétrico de 100 ml. Completar o volume com metanol e homogeneizar.
Transferir 1 ml da solução resultante para balão volumétrico de 100 ml, adicionar 1 ml de ácido clorídrico 0,1 M, completar o volume com metanol e homogeneizar. Preparar solução padrão na mesma concentração, utilizando os mesmos solventes. Medir as absorvâncias das soluções resultantes em 249 nm (V.2.14.-3) utilizando solução metanólica de ácido clorídrico 0,1 M para ajuste do zero. Calcular a quantidade de C8H9NO2 na solução oral, a partir das leituras obtidas. Alternativamente, realizar os cálculos considerando A(1%,1cm) = 880, em 249 nm.
B. Por cromatografia líquida de alta eficiência. Proceder conforme descrito em Cromatografia líquida de alta eficiência (V.2.17.4), utilizando cromatógrafo provido de detector ultravioleta a 243 nm, coluna de 300 mm de comprimento e 3,9 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octadecilsilano, fluxo da fase móvel de 1,5 ml/minuto.
Fase móvel: mistura de água e metanol (3:1).
Solução amostra: transferir um volume precisamente medido da solução oral, equivalente a 0,5 g de paracetamol, para balão volumétrico de 250 ml, completar o volume com a fase móvel e homogeneizar.
Transferir 5 ml desta solução para um segundo balão volumétrico de 250 ml, completar o volume com a fase móvel e homogeneizar.
Transferir 25 ml da solução anterior para balão volumétrico de 100 ml, completar o volume com a fase móvel, homogeneizar e filtrar.
Solução padrão: dissolver quantidade exatamente pesada de paracetamol padrão em metanol de modo a obter concentração final de 0,01 mg/ml.
O desvio padrão relativo das áreas do picos para injeções em replicatas não é superior a 2%.
Procedimento: injetar, separadamente, 10 μl das soluções padrão e amostra, registrar os cromatogramas e medir as áreas dos picos.
Calcular a quantidade de C8H9NO2 na solução amostra a partir das respostas obtidas para as soluções padrão e amostra.
EMBALAGEM E ARMAZENAMENTO
Conservar ao abrigo da luz.
ROTULAGEM
Observar a legislação vigente.
168
PERMANGANATO DE POTÁSSIO
Kalii permanganas
KMnO4 158,03 1023.09-8
Contém, no mínimo, 99,0% e, no máximo, 100,5% de KMnO4, em relação à substância dessecada.
Cuidado! Explosões perigosas podem ocorrer se posto em contato com substâncias orgânicas ou substâncias facilmente oxidáveis, tanto em solução como no estado seco.
DESCRIÇÃO
Caracteres físicos. Cristais violeta-escuros de brilho metálico azulado, inodoros, inalteráveis ao ar.
Solubilidade. Solúvel em água fria, facilmente solúvel em água fervente.
IDENTIFICAÇÃO
Responde às reações características do íon permanganato (V.3.1).
ENSAIOS DE PUREZA
Cloreto (V.3.2.1). Dissolver 0,75 g de permanganato de potássio em 25 ml de água, adicionar 3 ml de etanol 96%, aquecer por dois a três minutos e, em seguida, esfriar. Diluir para 30 ml com água e filtrar.
O filtrado é incolor. Diluir 10 ml do filtrado para 15 ml com água. No máximo 0,02% (200 ppm).
Sulfato (V.3.2.3). Diluir 12 ml do filtrado, obtido no ensaio de Cloreto, para 15 ml, com água. No máximo 0,05% (500 ppm).
Substâncias insolúveis em água. Dissolver 2 g da amostra em 150 ml de água, aquecer até ebulição. Filtrar imediatamente através de um filtro de vidro de média porosidade previamente tarado. Lavar o filtrado com três porções de 50 ml de água quente. Levar o filtro à estufa por 3 horas a 105 ºC. No máximo 0,2%.
Perda por dessecação (V.2.9). Determinar em dessecador sob pressão reduzida, sobre sílica-gel, por 18 horas, em 1 g de amostra. No máximo 0,5%.
DOSEAMENTO
Pesar, exatamente, cerca de 0,125 g da amostra e dissolver em 25 ml de água. Adicionar 2 ml de ácido sulfúrico previamente diluído com 5 ml de água e, em seguida, 50 ml de ácido oxálico 0,05 M, aquecer a solução a cerca de 80 ºC. Titular o excesso de ácido oxálico com permanganato de potássio 0,02 M SV até que seja produzida coloração rosa pálida persistente por 15 segundos. Cada ml de ácido oxálico 0,05 M SV equivale a 3,161 mg de KMnO4.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente
CLASSE TERAPÊUTICA
Anti-séptico tópico.
________________________________________________________
XII.3 SOLUÇÕES VOLUMÉTRICAS
Ácido oxálico 0,05 M SV
Especificação - Contém 6,45 g de ácido oxálico diidratado em água para 1 000 ml.
Padronização - Titular com permanganato de potássio 0,02 M SV recém-padronizado, conforme descrito em permanganato de potássio 0,02 M SV. Cada ml de permanganato de potássio equivale a 6,3025 mg de ácido oxálico diidratado.
Conservação - Em local protegido da luz.
Armazenagem - Em frascos providos de tampa esmerilhada.
Permanganato de potássio 0,02 M SV
Preparação - Dissolver cerca de 3,3 g de permanganato de potássio em 1 000 ml de água e ferver a solução por cerca de 15 minutos.
Deixar em repouso por, no mínimo, 2 dias em frasco bem-fechado.
Filtrar através de cadinho de gooch com camada de amianto.
Padronização - Pesar exatamente cerca de 0,2 g de oxalato de sódio, previamente dessecado a 110 ºC até peso constante e dissolver em 250 ml de água. Adicionar 7 ml de ácido sulfúrico, aquecer a cerca de 70 ºC e titular com a solução de permanganato de potássio até que uma coloração rosa pálida persistente por 15 segundos seja produzida.
A temperatura ao final da titulação não deve ser inferior a 60 ºC.
Cada ml de permanganato de potássio 0,02 M SV equivale a 6,7 mg de oxalato de sódio. Uma vez que o permanganato de potássio é reduzido em contato com substâncias orgânicas, deve-se operar com a aparelhagem totalmente de vidro ou outro material inerte adequado.
Conservação - Em local protegido da luz.
Armazenagem - Em frasco âmbar com tampa esmerilhada.
169
PROPILPARABENO
Propylis parahydroxibenzoas
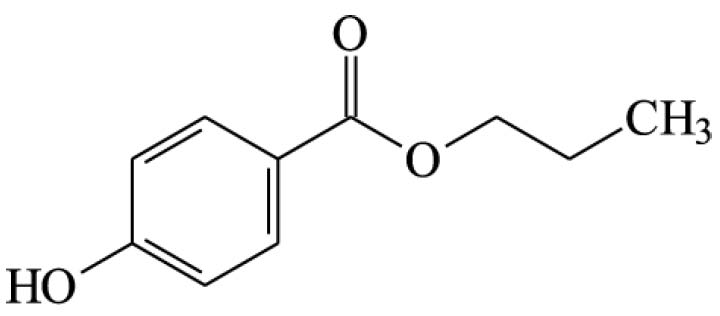
C10H12O3 180,20 1637.02-9
Éster propílico do ácido 4-hidroxibenzóico
Contém, no mínimo, 99,0% e, no máximo, 100,5% de C10H12O3.
DESCRIÇÃO
Caracteres físicos. Pó branco cristalino.
Solubilidade. Muito pouco solúvel em água e em água fervente, facilmente solúvel em metanol, etanol e éter etílico.
Constantes físico-químicas
Faixa de fusão (V.2.2): entre 96,0 oC e 99,0 oC.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14.-4.) da amostra dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de propilparabeno padrão, preparado de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14-3) na faixa de 230 a 280 nm, de uma solução a 0,0005% (p/V) em etanol, exibe máximo em 258 nm e a absorvância se encontra na faixa de 0,440 a 0,475.
ENSAIOS DE PUREZA
Aspecto da solução. A solução a 10% (p/V) em etanol é límpida e incolor.
Acidez. A 2 ml da solução descrita em Aspecto da solução, adicionar 3 ml de etanol, 5 ml de água isenta de dióxido de carbono e 0,1 ml de solução de verde de bromocresol SI. Não é necessário mais que 0,1 ml de solução de hidróxido de sódio 0,1 M SV para a viragem do indicador.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,1%.
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte, e ácido fórmico anidro-acetato de etila-cloreto de metileno (2:10:88), como fase móvel. Aplicar, separadamente, à placa, 5μl de cada uma das soluções, recentemente preparadas, descritas a seguir.
Solução (1): dissolver 0,1 g da amostra em metanol e diluir com o mesmo solvente para 5 ml.
Solução (2): diluir 1 ml da solução (1) para 100 ml com metanol.
Desenvolver o cromatograma. Remover a placa, deixar secar sob
corrente de ar quente e examinar sob luz ultravioleta (254 nm).
Qualquer mancha no cromatograma da solução (1), exceto a principal,
não é mais intensa do que a mancha obtida no cromatograma da
solução (2).
DOSEAMENTO
Transferir 2 g da amostra para um erlenmeyer provido de rolha esmerilhada, adicionar 40 ml de solução de hidróxido de sódio M SV.
Adaptar condensador de refluxo e aquecer cuidadosamente por 30 minutos. Titular o excesso de hidróxido de sódio com solução deácido sulfúrico 0,5 M SV determinando o ponto final potenciometricamente.
Efetuar ensaio em branco e fazer as correções necessárias.
Cada ml de hidróxido de sódio M SV equivale a 180,2 mg de C10H12O3.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados.
ROTULAGEM
Observar a legislação vigente.
CATEGORIA
Conservante.
170
SULFATO DE ATROPINA
Atropini sulfas
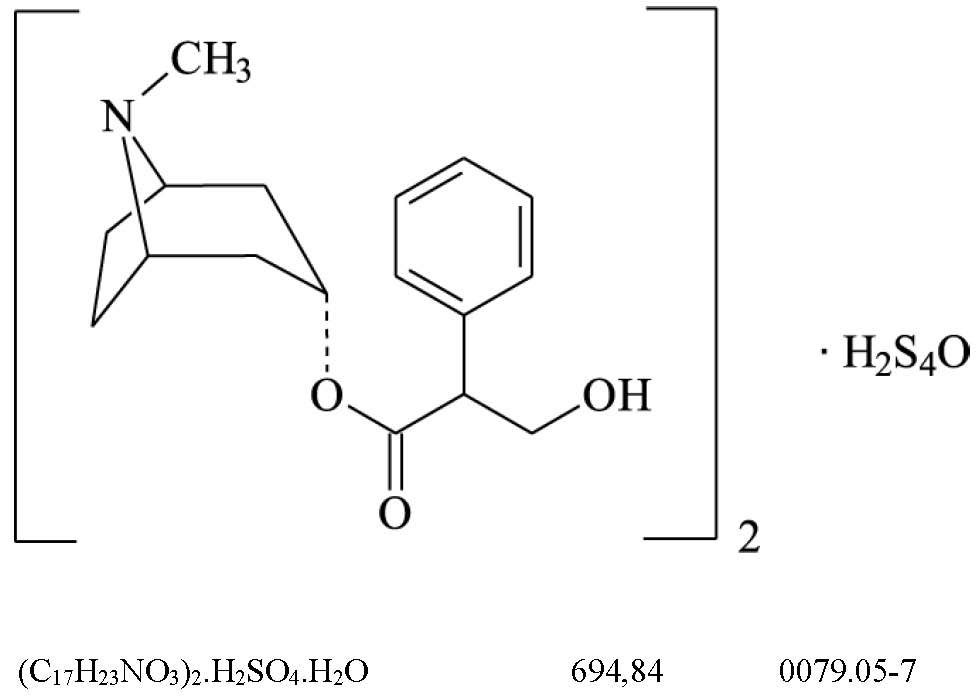
Sulfato de endo-(±)-α-(hidroximetil)benzeno-ácido acético 8-metil-8- azabiciclo[3.2.1]oct-3-il éster monoidratado
Contém, no mínimo, 98,5% e, no máximo, 101,0% de (C17H23NO3)2.H2SO4, em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Cristais incolores ou pó cristalino branco, inodoro, eflorescente ao ar seco, lentamente alterado pela luz.
Solubilidade. Muito solúvel em água, facilmente solúvel em etanol e em glicerina e praticamente insolúvel em éter etílico e clorofórmio.
Constantes físico-químicas
Ponto de fusão (V.2.2): não inferior a 187 oC Determinar imediatamente após dessecação da amostra a 120 oC por 4 horas.
IDENTIFICAÇÃO
A. Dissolver 50 mg da amostra em 25 ml de ácido clorídrico 0,01 M, adicionar 2 ml de hidróxido de sódio M e extrair com duas porções de 10 ml de éter etílico. Secar o extrato etéreo com sulfato de sódio anidro, filtrar, lavar com 5 ml de éter etílico e evaporar o filtrado em temperatura ambiente. Secar o resíduo sob sílica-gel, utilizando pressão reduzida. Paralelamente, realizar o mesmo procedimento utilizando 50 mg de sulfato de atropina padrão. O espectro de absorção no infravermelho (V.2.14.-4) do resíduo, disperso em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de sulfato de atropina padrão.
B. Proceder conforme descrito em Substâncias relacionadas. A mancha principal obtida com a solução (1) corresponde em cor, tamanho e intensidade àquela obtida com a solução (4).
C. A 1 mg da amostra, adicionar 0,2 ml de ácido nítrico fumegante e evaporar até secura em banho-maria. Dissolver o resíduo em 2 ml de acetona e adicionar 0,1 ml de solução de hidróxido de potássio a 3% (p/V) em metanol. Produz-se coloração violeta.
D. Dissolver alguns miligramas da amostra em 5 ml de água, acidificar com ácido clorídrico 2 M e adicionar 1 ml de solução de iodobismutato de potássio aquo-acético SR. Forma-se, imediatamente, precipitado alaranjado ou vermelho-alaranjado.
E. A 1 ml de solução aquosa a 5% (p/V) da amostra, adicionar 1 ml de água e 0,5 ml de solução de iodo 0,1 M. Forma-se precipitado pardo.
F. A solução aquosa a 5% (p/V) responde às reações do íon sulfato ( V. 3.1.1.- 5).
ENSAIOS DE PUREZA
pH (V.2.19). 4,5 a 6,2. Determinar em solução a 2% (p/V).
Substâncias relacionadas. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel G, como suporte, e mistura de acetona-água-amônia solução concentrada (90:7:3), como fase móvel. Aplicar, separadamente, à placa, 10 μl de cada uma das soluções descritas a seguir.
Solução (1): solução a 2% (p/V) da amostra em metanol.
Solução (2): solução a 0,02% (p/V) da amostra em metanol.
Solução (3): solução a 0,01% (p/V) da amostra em metanol.
Solução (4): solução a 2% (p/V) de sulfato de atropina padrão em metanol.
Desenvolver o cromatograma. Remover a placa, secar à temperatura de 100 oC a 105 oC, por 15 minutos. Deixar esfriar e nebulizar com solução de iodobismutato de potássio-ácido tartárico até aparecimento das manchas. Nenhuma mancha secundária obtida no cromatograma com a solução (1) é mais intensa que a mancha obtida com a solução (2) e não mais que uma mancha é mais intensa do que aquela obtida com a solução (3).
Apoatropina. Preparar solução a 0,1% (p/V) em ácido clorídrico 0,01 M. Medir a absorvância em 245 nm (V.2.14.-3), utilizando o mesmo solvente para ajuste do zero. O valor da absorvância é de, no máximo, 0,4 (0,5%).
Hiosciamina. Dissolver 1,25 g, exatamente pesados, em água, para volume final de 25 ml. Deteminar o ângulo de rotação (V.2.8) da solução, a 25 oC. A rotação observada, em graus, multiplicada por 200 e dividida pelo comprimento (em mm) do tubo polarimétrico usado, está entre -0,60 o e +0,05 o.
Água (V.2.20.1). No máximo 4,0%
Cinzas sulfatadas (V.2.10). No máximo 0,2%.
DOSEAMENTO
Proceder conforme descrito em titulação em meio não-aquoso (V.3.4.5.). Dissolver cerca de 1 g da amostra, exatamente pesada, em 50 ml de ácido acético glacial e titular com ácido perclórico 0,1 M SV, determinando o ponto final potenciometricamente. Realizar ensaio em branco e fazer as correções necessárias. Cada ml de ácido perclórico 0,1 M SV equivale a 67,682 mg de (C17H23NO3)2.H2SO4.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Midriático e adjuvante de anestésicos gerais.
170.1
SULFATO DE ATROPINA SOLUÇÃO INJETÁVEL
Solução estéril de sulfato de atropina em água para injetáveis. Contém, no mínimo, 90,0% e, no máximo, 110,0% da quantidade declarada de (C17H23NO3)2.H2SO4.H2O.
IDENTIFICAÇÃO
A. Utilizar volume da amostra equivalente a 10 mg de sulfato de atropina, adicionar 4 ml de hidróxido de sódio M e proceder conforme descrito no teste A de Identificação na monografia de sulfato de atropina, a partir de “extrair com duas porções de 10 ml de éter etílico...”.
B. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel G, como suporte, e mistura de clorofórmio- acetona-dietilamina (50:40:10), como fase móvel. Aplicar, separadamente, à placa, 5 μl de cada uma das soluções descritas a seguir.
Solução (1): evaporar um volume da solução injetável contendo o equivalente a 5 mg de sulfato de atropina, até secura, em banhomaria.
Triturar o resíduo com 1 ml de etanol, deixar em repouso e utilizar o sobrenadante.
Solução (2): solução a 0,5% (p/V) de sulfato de atropina padrão em etanol.
Desenvolver o cromatograma. Remover a placa, secar a 105 oC por 20 minutos. Deixar esfriar e nebulizar com solução de iodobismutato de potássio-ácido tartárico. A mancha principal obtida no cromatograma com a solução (1) corresponde em tamanho, cor e posição à mancha obtida no cromatograma com a solução (2).
C. Evaporar até secura volume da solução injetável equivalente a 1 mg de sulfato de atropina. Adicionar ao resíduo 0,2 ml de ácido nítrico fumegante e evaporar até secura em banho-maria. Forma-se resíduo amarelo. Após esfriar, adicionar 2 ml de acetona e 0,2 ml de solução a 3% (p/V) de hidróxido de potássio em metanol. Desenvolve- se coloração violeta.
D. Responde às reações do íon sulfato (V.3.1.1.-5).
CARACTERÍSTICAS
Determinação de volume (V.1.2). Cumpre o teste.
pH (V.2.19). 3,0 a 6,5.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1). Cumpre o teste.
Endotoxinas bacterianas (V.5.1.9). No máximo 55,6 UE/mg de sulfato de atropina.
DOSEAMENTO
Proceder conforme descrito em Cromatografia líquida de alta eficiência (V.2.17.4), utilizando cromatógrafo provido de detector ultravioleta a 254 nm; coluna de 300 mm de comprimento e 3,9 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octadecilsilano (5 μm ou 10 μm), mantida à temperatura ambiente;
fluxo da fase móvel de 2 ml/minuto.
Tampão acetato: dissolver o equivalente a 0,05 mol de acetato de sódio em água, adicionar 2,9 ml de ácido acético glacial e completar o volume com água para 1 000 ml.
Fase móvel: transferir 5,1 g de sulfato ácido de tetrabutilamônio para balão volumétrico de 1 000 ml, adicionar 50 ml de acetonitrila e completar o volume com tampão acetato. Ajustar o pH para 5,5 ± 0,1 com hidróxido de sódio 5 M.
Solução amostra: transferir volume do medicamento equivalente a cerca de 2 mg de sulfato de atropina monoidratado para balão volumétrico de 25 ml, completar o volume com água e homogeneizar.
Solução padrão: dissolver quantidade exatamente pesada de sulfato de atropina padrão e diluir com água de modo a obter concentração equivalente a 80 μg/ml de sulfato de atropina diidratado.
Solução de resolução: diluir um volume de solução aquosa de ácido p-hidroxibenzóico a 2,5 μg/ml com quatro volumes da solução padrão.
Injetar 100 μl.
O tempo de retenção do ácido p-hidroxibenzóico é cerca de 1,6 vezes superior ao do sulfato de atropina. A resolução entre os picos doácido p-hidroxibenzóico e do sulfato de atropina não é inferior a 2,2.
O desvio padrão relativo das áreas de replicatas dos picos obtidos não é superior a 1,5%.
Procedimento: injetar, separadamente, 100 μl das soluções padrão e amostra, registrar os cromatogramas e medir as áreas dos picos.
Calcular a quantidade de (C17H23NO3)2.H2SO4.H2O na solução injetável a partir das respostas obtidas para as soluções padrão e amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes de dose única ou dose múltipla, preferencialmente de vidro tipo I.
ROTULAGEM
Observar a legislação vigente.
________________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Solução de iodobismutato de potássio-ácido tartárico
Preparação - Dissolver 10 g de ácido tartárico em 40 ml de água e adicionar 0,85 g de subnitrato de bismuto. Agitar durante uma hora, adicionar 20 mlde solução de iodeto de potássio a 40% (p/V) e homogeneizar. Deixar em repouso durante 24 horas e filtrar. Dissolver 10 g de ácido tartárico em 50 ml de água, adicionar 5 ml da solução anterior e homogeneizar.
171
ZIDOVUDINA
Zidovudinum
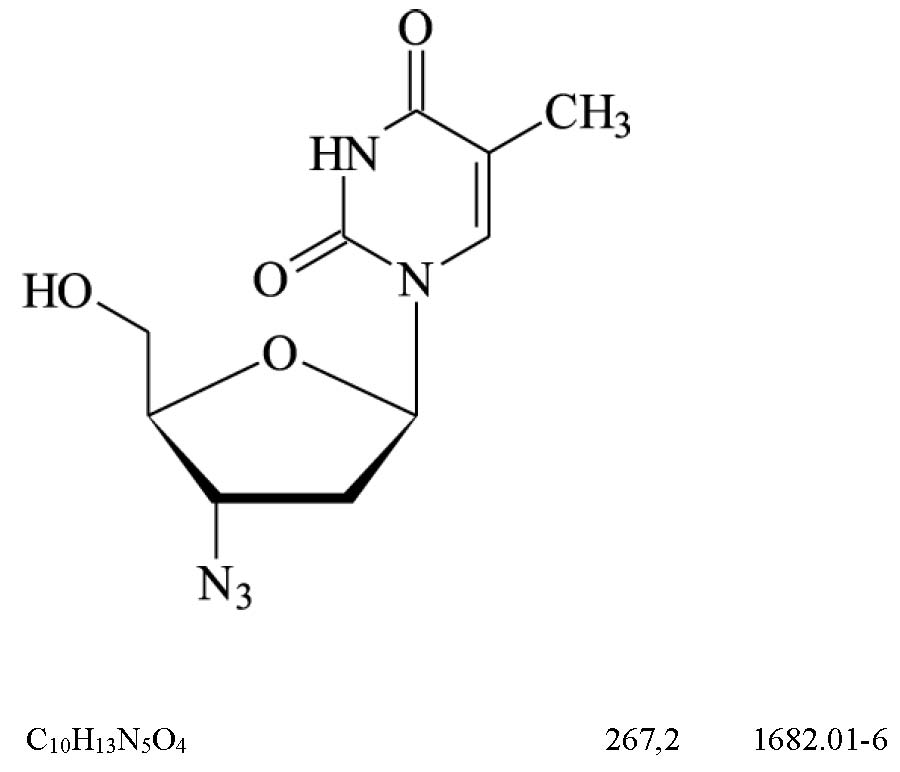
1-(3-Azido-2,3-didesoxi-β-D-ribofuranosil)-5-metilpirimidino- 2,4(1H,3H)-diona
Contém, no mínimo, 97,0% e, no máximo, 102,0% de C10H13N5O4, em relação à substância dessecada.
DESCRIÇÃO
Caracteres físicos. Pó branco ou acastanhado. Apresenta polimorfismo.
Solubilidade. Levemente solúvel em água, solúvel em etanol.
Constantes físico-químicas
Ponto de fusão (V.2.2): funde a aproximadamente 124 oC.
Poder rotatório específico (V.2.8): +60,5o a +63,0o, a 25 oC, em relação à substância dessecada. Determinar em solução a 1% (p/V) em etanol.
IDENTIFICAÇÃO
A. O espectro de absorção no infravermelho (V.2.14-4) da amostra dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de zidovudina padrão, preparado de maneira idêntica.
B. Proceder conforme indicado em Poder rotatório específico.
ENSAIOS DE PUREZA
Limpidez e cor da solução (IV.-3 e V.2.12). Dissolver 0,5 g em 50 ml de água, aquecendo se necessário. A solução não é mais intensamente corada do que a solução padrão de cor obtida pela mistura de 1 ml da solução padrão de cor G com 7 ml de água.
Perda por dessecação (V.2.9). Determinar em 1 g de amostra, em estufa, entre 100 oC e 105 oC, até peso constante. No máximo 1%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,25%.
Substâncias relacionadas
A. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel GF254, como suporte e mistura de metanol e diclorometano (10:90), como fase móvel. Aplicar, separadamente,à placa, 10 μl de cada uma das soluções, recentemente preparadas, descritas a seguir.
Solução (1): dissolver 0,2 g de amostra em metanol e diluir para 10 ml com o mesmo solvente.
Solução (2): dissolver em metanol 20 mg de timina padrão, 20 mg da impureza de zidovudina A (1-[(2R,5S)-5-hidroximetil-2,5-diidro-2-furil]- 5-metilpirimidino-2,4(1H,3H)-diona), 20 mg de trifenilmetanol, adicionar 1 ml da solução (1) e diluir para 100 ml com metanol.
Solução (3): diluir 5 ml da solução (2) para 10 ml com metanol.
Desenvolver o cromatograma. Aguardar a ascensão do solvente até 12 cm acima da linha de aplicação. Remover a placa, deixar secar ao ar por 5 minutos e examinar sob luz ultravioleta (254 nm). A mancha correspondente à impureza de zidovudina A não é mais intensa do que a mancha correspondente no cromatograma obtido com a solução (3) (no máximo 0,5%) e qualquer mancha, com exceção da principal e as manchas correspondentes à impureza de zidovudina e timina não são mais intensas do que a mancha correspondente à zidovudina no cromatograma obtido com a solução (3) (no máximo 0,5%). Nebulizar a placa com solução de vanilina a 1% (p/V) em ácido sulfúrico.
No cromatograma obtido com a solução (1), qualquer mancha correspondente ao trifenilmetanol não é mais intensa do que a mancha correspondente no cromatograma obtido com a solução (3) (no máximo 0,5%). O teste é válido se o cromatograma obtido com a solução (2) apresentar quatro manchas claramente separadas, correspondentesà timina padrão, à impureza de zidovudina A, à zidovudina e ao trifenilmetanol, em ordem crescente de fator de retenção (Rf).
B. Proceder conforme descrito no Doseamento. Injetar, separadamente, 10 μl de cada umas das soluções: solução (1), solução (4), solução (6) e solução (7). Continuar a cromatografia por 1,5 vezes o tempo de retenção do pico principal no cromatograma obtido com a solução (1). As substâncias são eluídas na seguinte sequência: timina, zidovudina e impureza de zidovudina B. No cromatograma obtido com a solução (1), a área de qualquer pico correspondente à timina não é maior do que a área do pico no cromatograma obtido com a solução (4) (no máximo 2%). A área de qualquer pico correspondenteà impureza de zidovudina B não é maior do que a área do pico correspondente no cromatograma obtido com a solução (6) (no máximo 1%). A área de qualquer outro pico, com exceção do principal, não é maior do que a área do pico no cromatograma obtido com a solução (7) (no máximo 0,5%). A soma das áreas de todos os picos, com exceção do principal, obtidos no cromatograma com solução (1), não é maior do que seis vezes a área do pico obtida com solução (7) (no máximo 3%). Desprezar qualquer pico com área menor do que 10% da área do pico obtido no cromatograma obtido com a solução (7).
Metais pesados (V.3.2.3.-3 - Método IV). Determinar em 1 g de amostra. Utilizar 2 ml de solução padrão de chumbo (10 ppm). No máximo 0,002% (20 ppm).
DOSEAMENTO
Proceder conforme descrito em Cromatografia líquida de alta eficiência (V.2.17.4), utilizando cromatógrafo provido de detector ultravioleta, a 265 nm; coluna cromatográfica de 250 mm de comprimento e 4,6 mm de diâmetro interno, empacotada com sílica quimicamente ligada a octadecilsilano (5 μm); fluxo da fase móvel de 1,2 ml/minuto.
Fase móvel: mistura de metanol-água (20:80).
Solução (1): dissolver 50 mg da amostra na fase móvel e diluir para 50 ml com o mesmo solvente.
Solução (2): diluir 10 ml da solução (1) para 50 ml com a fase móvel.
Solução (3): dissolver 10 mg de zidovudina padrão na fase móvel e diluir para 50 ml com o mesmo solvente.
Solução (4): dissolver 10 mg de timina padrão em metanol e diluir para 50 ml com o mesmo solvente. Diluir 5 ml desta solução para 50 ml com a fase móvel.
Solução (5): dissolver 5 mg da impureza de zidovudina B (1-(3-cloro- 2,3-didesoxi-β-D-ribofuranosil)-5-metilpirimidino-2,4(1H,3H)-diona, em 25 ml da solução (3) e diluir para 50 ml com a fase móvel.
Solução (6): diluir 5 ml da solução (5) para 50 ml com a fase móvel.
Solução (7): diluir 0,25 ml da solução (1) para 50 ml com a fase móvel.
O fator de cauda não deve ser maior que 1,5. A resolução entre os picos da zidovudina e da impureza de zidovudina B não é menor que
1,4. O desvio padrão relativo das áreas das replicatas dos picos obtidos não é maior que 2%.
Procedimento: injetar, separadamente, 10 μl das soluções (2) (3) e (5), registrar os cromatogramas e medir as áreas dos picos. Calcular o teor de C10H13N5O4 na amostra, a partir das respostas obtidas para as soluções (2) e (3).
ARMAZENAMENTO
Armazenar em recipientes bem-fechados, protegidos da luz.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Anti-retroviral.
TEXTOS A SEREM INCLUÍDOS NA PARTE I
V.2.23 ESPECTROFOTOMETRIA DE EMISSÃO ATÔMICA
Espectrofotometria de emissão atômica é o método que permite determinar a concentração de um elemento numa substância pela medida da intensidade de uma das linhas de emissão do vapor do elemento ao ser atomizado. A determinação é feita no comprimento de onda correspondente a esta linha de emissão.
Equipamento
Consiste de um atomizador (chama, plasma, arco, entre outros), do monocromador e do detector. Se o atomizador é a chama, o solvente de escolha para o preparo da solução amostra e soluções de referênciaé a água. Os solventes orgânicos podem ser usados, desde que se assegure que o solvente não interfira na estabilidade da chama.
OPERAÇÃO
O equipamento deve ser operado de acordo com as instruções do fabricante e no comprimento de onda prescrito. Ajusta-se o zero do equipamento com o solvente injetado no equipamento. Em seguida, injeta-se a solução de referência mais concentrada e a sensibilidade do aparelho é ajustada para obter a sensibilidade desejada. As determinações são feitas por comparação com soluções de referência, contendo concentrações conhecidas do elemento em análise. As determinações podem ser feitas pelo Método de Calibração Direta (Método
I) ou pelo Método de Adição Padrão (Método II).
Método I (Calibração direta)
Preparar ao menos três soluções de referência do elemento a ser dosado, abrangendo a faixa de concentrações recomendada pelo fabricante do aparelho para o elemento em análise. Todos os reagentes empregados no preparo da solução amostra devem ser igualmente incluídos, nas mesmas concentrações às soluções de referência. Após a calibração do aparelho com solvente, injetar, três vezes, cada uma das soluções de referência e, após a estabilização da leitura, registrar o resultado, lavando o sistema com o solvente após cada injeção.
Traçar a curva de calibração, plotando a média das leituras de cada grupo de três, com a respectiva concentração. Preparar a solução da substância a ser dosada conforme indicado na monografia, ajustando sua concentração para que esta fique dentro da faixa das concentrações das soluções de referência. Injetar a solução amostra no aparelho, registrar a leitura e lavar o sistema com o solvente. Repetir esta seqüência duas vezes e, adotando a média de três leituras, determinar a concentração do elemento pela curva de concentração.
Método II (Adição padrão)
Adicionar a cada um de, ao menos, três balões volumétricos similares, volumes iguais de solução da substância a ser dosada, preparada conforme indicado na monografia. Juntar a todos os balões, com exceção de um, volumes medidos da solução de referência especificada, de modo a obter uma série de soluções contendo quantidades crescentes do elemento sob análise. Diluir convenientemente o volume de cada balão com água. Após calibrar o espectrofotômetro com água, como indicado acima, registrar três vezes as leituras de cada solução. Transferir os resultados das leituras e as concentrações correspondentes para gráfico cujos eixos interceptem em zero de elemento adicionado e zero de leitura. Extrapolar a linha reta que une os pontos até a interceptação do eixo de concentrações. A distância entre este ponto e a intersecção dos eixos representa a concentração do elemento dosado na amostra.
V.3.2.7 ENSAIO-LIMITE PARA CÁLCIO
A 0,2 ml da solução padrão alcoólica de cálcio 100 ppm, adicionar 1 ml de oxalato de amônio SR. Aguardar 1 minuto e adicionar mistura de 1 ml de ácido acético diluído e 15 ml da solução amostra preparada como descrito na monografia. Agitar. Preparar o padrão da mesma maneira, utilizando mistura de 10 ml da solução padrão de cálcio 10 ppm, 1 ml de ácido acético diluído e 5 ml de água.
Após 15 minutos, qualquer opalescência da preparação amostra não é mais intensa do que a obtida com a preparação padrão.
Solução padrão de cálcio 400 ppm - Dissolver em água 1,000 g de carbonato de cálcio e 23 ml de ácido clorídrico M. Completar o volume para 100 ml com água. Imediatamente antes do uso, diluir 10 ml desta solução para 100 ml com água.
Solução padrão de cálcio 100 ppm - Dissolver em água 0,624 g de carbonato de cálcio e 3 ml de ácido acético. Completar o volume comágua para 250 ml. Imediatamente antes do uso, diluir 10 ml desta solução para 100 ml com água.
Solução padrão alcoólica de cálcio 100 ppm - Dissolver em água 2,5 g de carbonato de cálcio e 12 ml de ácido acético. Completar o volume com água para 1 000 ml. Imediatamente antes do uso, diluir 10 ml desta solução para 100 ml com etanol.
Solução padrão de cálcio 10 ppm - Dissolver em água 0,624 g de carbonato de cálcio e 3 ml de ácido acético. Completar o volume comágua para 250 ml. Imediatamente antes do uso, diluir 10 ml desta solução para 1 000 ml com água.
V.3.2.8 ENSAIO-LIMITE PARA MAGNÉSIO
Medir 10 ml da solução amostra e adicionar 0,1 g de tetraborato sódico. Ajustar o pH para a faixa compreendida entre 8,8 e 9,2 com ácido clorídrico SR ou hidróxido de sódio SR. Transferir para ampola de decantação e extrair 2 vezes, agitando por 1 minuto cada vez, com 5 ml da solução de hidroxiquinolina a 0,1% (p/V) em clorofórmio.
Deixar decantar, separar e descartar a camada orgânica. Adicionar à camada aquosa 0,4 ml de butilamina e 0,1 ml de trietanolamina.
Ajustar o pH da solução para a faixa de 10,5 a 11,5, se necessário.
Adicionar 4 ml da solução de hidroxiquinolina a 0,1% (p/V) em clorofórmio, agitar por 1 minuto e deixar separar as camadas. Utilizar a camada inferior para a comparação. Preparar solução padrão de magnésio da mesma maneira, utilizando mistura de 1 ml da solução padrão de magnésio (10 ppm de Mg) e 9 ml de água.
Qualquer coloração obtida na preparação amostra não é mais intensa do que a coloração da preparação padrão.
Solução padrão de magnésio 100 ppm - Dissolver 1,010 g de sulfato de magnésio heptaidratado em água e completar o volume para 100 ml com água. Diluir 10 ml desta solução para 100 ml com água.
Solução padrão de magnésio 10 ppm - Dissolver 1,010 g de sulfato de magnésio heptaidratado em água e completar o volume a 100 ml comágua. Diluir 10 ml desta solução para 1 000 ml com água.
________________________________________________________
XII.2. REAGENTES E SOLUÇÕES REAGENTES
Butilamina
Fórmula e massa molecular - C4H11 N - 73,1
Descrição - Líquido incolor, miscível com água, etanol e éter etílico.
Características físicas - Faixa de ebulição: aproximadamente 78 oC.
Informação adicional - Destilar e utilizar dentro de um mês.
Sulfato de magnésio heptaidratado
Fórmula e massa molecular - MgSO4.7H2O - 246,5
Descrição - Pó branco cristalino ou cristais incolores brilhantes, solúveis em água, muito solúveis em água fervente, praticamente insolúveis em etanol.
Trietanolamina
Sinonímia - 2,2',2''-nitrilotrietanol
Fórmula e massa molecular - C6H15NO3 - 149,2
Descrição - Líquido incolor, viscoso, muito higroscópico, que se torna marrom pela exposição ao ar. Miscível com água, acetona, etanol e metanol.
Características físicas - Densidade: aproximadamente 1,13.
Conservação - Armazenar em recipientes hermeticamente fechados ao abrigo da luz.
V.3.2.9 ENSAIO-LIMITE PARA MAGNÉSIO E METAIS ALCALINO-TERROSOS
A 200 ml de água adicionar 0,1 g de cloridrato de hidroxilamina, 10 ml tampão cloreto de amônio pH 10,0, 1 ml de solução de sulfato de zinco 0,1 M e cerca de 15 mg de negro de eriocromo T, triturado.
Aquecer a cerca de 40 oC. Titular com edetato dissódico 0,01 M SV até a coloração violeta mudar para azul. Adicionar a esta solução quantidade prescrita da amostra em exame, dissolvida em 100 ml deágua ou preparada de modo descrito na monografia. Se a coloração da solução mudar para violeta, titular com edetato dissódico 0,01 M SV até a viragem para azul.
O volume da solução do edetato dissódico 0,01 M SV utilizado na segunda titulação não excede ao estabelecido na monografia.
_________________________________________________
XII.2 REAGENTES E SOLUÇÕES REAGENTES
Cloridrato de hidroxilamina
Fórmula e massa molecular - NH4ClO - 69,5
Descrição - Pó branco cristalino, muito solúvel em água, solúvel em etanol.
V.3.2.10 ENSAIO-LIMITE PARA ALUMÍNIO
Introduzir num funil de separação a quantidade prescrita da solução da amostra e extrair com duas porções de 20 ml cada e, em seguida com uma porção de 10 ml da solução de hidroxiquinolina a 0,5% (p/V) em clorofórmio. Diluir os extratos clorofórmicos combinados para 50 ml com clorofórmio.
Preparar o padrão de maneira idêntica, utilizando quantidade prescrita da solução padrão.
Preparar um branco da maneira idêntica, utilizando solução prescrita.
Medir a intensidade da fluorescência (V.2.15) da solução amostra (I1), do padrão (I2) e do branco (I3) utilizando o feixe de excitação em 392 nm e um filtro secundário com a faixa de transmissão centrada em 518 nm ou com o monocromador ajustado para transmitir neste comprimento de onda.
A fluorescência (I1 - I3) da solução amostra não deve ser maior do que a do padrão (I2 - I3)
Solução padrão de alumínio 200 ppm - Dissolver em água 0,352 g de sulfato de alumínio e potássio dodecaidratado. Adicionar 10 ml deácido sulfúrico diluído e diluir com água para 100 ml.
Solução padrão de alumínio 10 ppm - Dissolver 1,39 g de nitrato de alumínio nonaidratado em água e completar o volume para 100 ml.
No momento do uso diluir 10 ml desta solução com água para 1 000 ml.
Solução padrão de alumínio 2 ppm - Dissolver em água 0,352 g de sulfato de alumínio e potássio dodecaidratado. Adicionar 10 ml deácido sulfúrico diluído e diluir com água para 100 ml. No momento do uso diluir 10 ml desta solução para 1 000 ml.
_________________________________________________
XII.2 REAGENTES E SOLUÇÕES REAGENTES
Ácido sulfúrico diluído
Especificação - Contém 9,8 % (p/V) de H2SO4
Preparação - Adicionar 5,5 ml de ácido sulfúrico a 60 ml de água.
Esfriar e diluir com água para 100 ml.
Hidroxiquinolina
Sinonímia - 8-hidroxiquinolina
Fórmula e massa molecular - C9H7NO - 145,2
Descrição - Pó cristalino, branco ou levemente amarelado, pouco solúvel em água, muito solúvel em acetona, em etanol e em soluções diluídas de ácidos minerais.
Características físicas - Faixa de fusão: aproximadamente 75 oC.
Nitrato de alumínio nonaidratado
Fórmula e massa molecular - Al(NO3)3.9H2O - 375,1
Descrição - Cristais deliquescentes, muito solúveis em água e em etanol, muito pouco solúveis em acetona.
Conservação - Em recipientes hermeticamente fechados.
Sulfato de alumínio e potássio dodecaidratado
Sinonímia - Alúmen de potássio
Fórmula e massa molecular - AlK(SO4)2.12H2O - 474,4
Descrição - Pó granular ou massa incolor, transparente, muito solúvel em água fervente, solúvel em glicerina, praticamente insolúvel em etanol.
Conservação - Em recipientes bem fechados.
V.3.2.11 ENSAIO-LIMITE PARA FOSFATOS
A 100 ml da solução preparada conforme descrito na monografia, adicionar 4 ml de reagente sulfomolíbdico e agitar. Adicionar 0,1 ml de solução de cloreto estanoso SR e agitar. Proceder do mesmo modo utilizando 2 ml da solução padrão de fosfato 5 ppm e 98 ml de água.
Aguardar 10 minutos e comparar as cores utilizando 20 ml de cada preparação.
A coloração da preparação amostra não é mais intensa do que a da preparação padrão.
Solução padrão de fosfato 5 ppm - Dissolver 0,716 g de fosfato monobásico de potássio em 1 000 ml de água. No momento do uso diluir 10 ml desta solução para 1 000 ml com água.
________________________________________________
XII.2 REAGENTES E SOLUÇÕES REAGENTES
Reagente sulfomolíbdico
Preparação - Dissolver com aquecimento 2,5 g de molibdato de amônio em 20 ml de água. Diluir 28 ml de ácido sulfúrico em 50 ml deágua e esfriar. Misturar as duas soluções e diluir para 100 ml comágua.
V.4.3.1 DETERMINAÇÃO DE METANOL E 2-PROPANOL EM EXTRATOS FLUIDOS
Proceder a destilação do extrato conforme descrito em Determinação de etanol (V.3.4.8.1). Examinar o destilado por cromatografia a gás (V.2.17.5), utilizando cromatógrafo provido de detector de ionização de chama, coluna cromatográfica de vidro, com 2 m de comprimento e 2 mm de diâmetro interno empacotada com copolímero de etilvinilbenzeno/divinilbenzeno, partículas de 125μ m a 150μ m, e nitrogênio para cromatografia como gás de arraste, com fluxo de 30 ml/min. Manter a temperatura da coluna em130 oC, a temperatura do injetor em 200 oC e a temperatura do detector em 220 oC.
Solução do padrão interno
Preparar solução contendo 2,5 % (V/V) de 1-propanol em água.
Solução amostra
Adicionar a volume determinado de destilado 2 ml da solução de padrão interno. Ajustar o teor de etanol para 10 % (V/V) pela diluição de 50 ml de água ou adição de etanol a 90 % (V/V).
Solução padrão
Preparar 50 ml de solução contendo 2 ml da solução do padrão interno, 10 % de etanol a 90 % (V/V), 0,05 % de 2-propanol (V/V) e quantidade suficiente de metanol anidro, de modo a obter concentração final de metanol de 0,05 % (V/V).
Procedimento: Injetar separadamente μ l de cada uma das soluções.
Calcular os teores de metanol e 2-propanol com referência à amostra original.
TEXTO QUE SUBSTITUI O PUBLICADO, ANTERIORMENTE, NA PARTE I
CONTEÚDO
I PREFÁCIO
II HISTÓRICO
III COMISSÃO PERMANENTE DE REVISÃO DA FARMACOPÉIA BRASILEIRA
IV GENERALIDADES
V MÉTODOS DE ANÁLISE
V.1 PROCEDIMENTOS TÉCNICOS APLICADOS A MEDICAMENTOS
V.1.1 Determinação de peso em formas farmacêuticas
V.1.2 Determinação de volume em formas farmacêuticas
V.1.3 Determinação de resistência mecânica em comprimidos
V.1.3.1 Dureza
V.1.3.2 Friabilidade
V.1.4 Teste de desintegração
V.1.4.1 Determinação do tempo de desintegração para comprimidos e cápsulas
V.1.4.2 Determinação do tempo de desintegração de supositórios,óvulos e comprimidos vaginais
V.1.5 Determinação do tempo de dissolução para comprimidos e cápsulas
V.1.6 Uniformidade de doses unitárias
V.2 MÉTODOS FÍSICOS E FÍSICO-QUÍMICOS
V.2.1 Determinação da massa
V.2.2 Determinação da temperatura e faixa de fusão
V.2.3 Determinação da temperatura de ebulição e faixa de destilação
V.2.4 Determinação da temperatura de congelamento
V.2.5 Determinação da densidade de massa e densidade relativa
V.2.6 Determinação do índice de refração
V.2.7 Determinação da viscosidade
V.2.8 Determinação do poder rotatório e do poder rotatório específico
V.2.9 Determinação da perda por dessecação
V.2.10 Determinação de cinzas sulfatadas (resíduo de incineração)
V.2.11 Determinação da granulometria de pós
V.2.12 Cor de líquidos
V.2.13 Espectrofotometria de absorção atômica
V.2.14 Espectrofotometria de absorção no ultravioleta, visível e infravermelho
V.2.15 Espectrofotometria de fluorescência
V.2.16 Turbidimetria e nefelometria
V.2.17 Cromatografia
V.2.17.1 Cromatografia em camada delgada
V.2.17.2 Cromatografia em papel
V.2.17.3 Cromatografia em coluna
V.2.17.4 cromatografia líquida de alta pressão
V.2.17.5 Cromatografia a gás
V.2.17.6 Material para cromatografia
V.2.18 Polarografia
V.2.19 Determinação do pH
V.2.20 Determinação de água
V.2.20.1 Método volumétrico
V.2.20.2 Método da destilação azeotrópica
V.2.20.3 Método gravimétrico
V.2.21 Análise de solubilidade por fases
V.2.22 Eletroforese
V.2.23 Espectrofotometria de emissão atômica
V.3 MÉTODOS QUÍMICOS
V.3.1 Reações de identificação
V.3.1.1 Íons, grupos e funções
Acetato
Acetila
Alcalóide
Alumínio, íon
Amina aromática primária
Amônia e amina alifática volátil
Amônio, íon
Antimônio III, íon
Arsênio
Barbitúrico sem substituinte no nitrogênio
Bário, íon
Benzoato
Bicarbonato
Bismuto, íon
Bissulfito
Borato
Brometo
Cálcio, íon
Carbonato
Chumbo, íon
Cianeto
Citrato
Clorato
Cloreto
Cobre II, íon
Éster
Ferro
Férrico, íon
Ferroso, íon
Fosfato (ou ortofosfato)
Hipofosfito
Iodeto
Lactato
Lítio, íon
Magnésio, íon
Mercúrio
Mercúrio II, íon
Mercúrio III, íon
Nitrato
Nitrito
Oxalato
Permanganato
Peróxido
Potássio, íon
Prata, íon
Salicilato
Sódio, íon
Succinato
Sulfato
Sulfito
Tartarato
Tiocianato
Tiossulfato
Xantina
Zinco, íon
V.3.1.2 Identificação de esteróides por cromatografia em camada delgada V.3.1.3 Pesquisa de esteróides estranhos por cromatografia em camada delgada
V.3.1.4 Pesquisa de substâncias relacionadas a sulfonamidas por cromatografia em camada delgada
V.3.1.5 Identificação de fenotiazinas por cromatografia em camada delgada
V.3.1.6 Pesquisa de impurezas relacionadas a fenotiazinas por cromatografia em camada delgada
V.3.2 Ensaios-limite para impurezas inorgânicas
V.3.2.1 Ensaio-limite para cloretos
V.3.2.2 Ensaio-limite para sulfatos
V.3.2.3 Ensaio-limite para metais pesados
V.3.2.4 Ensaio-limite para ferro
V.3.2.5 Ensaio-limite para arsênio
V.3.2.6 Ensaio-limite para amônia
V.3.2.7 Ensaio-limite para cálcio
V.3.2.8 Ensaio-limite para magnésio
V.3.2.9 Ensaio-limite para magnésio e metais alcalino-terrosos
V.3.2.10 Ensaio-limite para alumínio
V.3.2.11 Ensaio-limite para fosfatos
V.3.3 Determinações em gorduras e óleos
V.3.3.1 Determinação da densidade relativa
V.3.3.2 Determinação da temperatura de fusão
V.3.3.3 Determinação da temperatura de solidificação
V.3.3.4 Determinação do índice de refração
V.3.3.5 Determinação do poder rotatório
V.3.3.6 Determinação de água e sedimentos
V.3.3.7 Determinação do índice de acidez
V.3.3.8 Determinação do índice de saponificação
V.3.3.9 Determinação do índice de ésteres
V.3.3.10 Determinação do índice de iodo
V.3.3.11 Determinação do índice de peróxidos
V.3.3.12 Determinação do índice de hidroxila
V.3.3.13 Determinação matéria insaponificável
V.3.4 Ensaios
V.3.4.1 Titulações por diazotação
V.3.4.2 Determinação de nitrogênio pelo método de Kjeldahl
V.3.4.2.1 Macrodeterminação (Método I)
V.3.4.2.2 Semi-microdeterminação (Método II)
V.3.4.3 Método de combustão em frasco de oxigênio
V.3.4.4 Titulações complexométricas
Alumínio
Bismuto
Cálcio
Chumbo
Magnésio
Zinco
V.3.4.5 Titulações em meio não-aquoso
Titulação de substâncias de caráter básico
Titulação de sais de ácidos halogenados
Titulação de substâncias de caráter ácido
V.3.4.6 Determinação de metoxila
V.3.4.7 Determinação do dióxido de enxofre
V.3.4.8 Determinação do álcool
V.3.4.8.1 Método por destilação
V.3.4.8.2 Método por cromatografia a gás
V.3.4.9 Análise de aminoácidos
V.4 MÉTODOS DE FARMACOGNOSIA
V.4.1 Preparo de material vegetal para observação e estudos histológicos
V.4.1.1 Amostragem qualitativa
V.4.1.2 Preparação do material para análise microscópica
V.4.2 Métodos de análise de drogas vegetais
V.4.2.1 Amostragem
V.4.2.2 Determinação de matéria estranha
V.4.2.3 Determinação de água em drogas vegetais
V.4.2.4 Determinação de cinzas totais
V.4.2.5 Determinação de cinzas insolúveis em ácido
V.4.2.6 Determinação de óleos essenciais em drogas vegetais
V.4.2.7 Determinação de óleos fixos
V.4.2.8 Determinação de cineol
V.4.2.9 Determinação do índice de espuma
V.4.2.10 Determinação de substâncias extraíveis por álcool (extrato alcoólico)
V.4.2.11 Determinação do índice de amargor
V.4.2.12 Determinação da atividade hemolítica
V.4.2.13 Determinação do índice de intumescência
V.4.3 Métodos de análise de extratos vegetais
V.4.3.1 Determinação de metanol e 2-propanol em extratos fluidos
V.5 MÉTODOS BIOLÓGICOS
V.5.1 Testes de segurança biológica
V.5.1.1 Esterilidade
V.5.1.2 Pirogênios
V.5.1.3 Toxicidade
V.5.1.4 Substâncias vasodepressoras
V.5.1.5 Histamina
V.5.1.6 Contagem de microrganismos viáveis em produtos que não necessitam cumprir com o Teste de esterilidade
V.5.1.7 Método geral para pesquisa e identificação de patógenos
V.5.1.7.1 Enriquecimento não-seletivo
- Substâncias solúveis na água
- Substâncias oleosas miscíveis em água
- Substâncias solúveis em miristato de isopropila
- Gelatina
- Substâncias insolúveis ou parcialmente solúveis na água
V.5.1.7.2 Fase seletiva e métodos de confirmação
- Pseudomonas aeruginosa
- Staphylococcus aureus
- Salmonella sp.
- Escherichia coli
V.5.1.7.3 Descrição dos meios de cultura e reagentes
V.5.1.7.4 Esterilização e acondicionamento dos meios de cultura
V.5.1.7.5 Capacidade seletiva e nutritiva dos meios de cultura e validação do teste para pesquisa e identificação de patógenos
V.5.1.8 Substâncias pressoras
V.5.1.9 Endotoxinas bacterianas
V.5.2 Ensaios
V.5.2.1 Ensaio biológico de oxitocina
Método A: hipotensão arterial em frango
Método B: contração do útero de rata in vitro
V.5.2.2 Ensaio biológico de corticotrofina
Método A: subcutâneo
Método B: intravenoso
V.5.2.3 Ensaio biológico de insulina
Método A: convulsão em camundongos
Método B: glicose sangüínea em coelhos
Método C: glicose sanguínea em camundongos
V.5.2.4 Duração do efeito da insulina
V.5.2.5 Ensaio biológico de glucagon
V.5.2.6 Ensaio biológico de heparina
V.5.2.7 Ensaio biológico de sulfato de protamina
V.5.2.8 Ensaio biológico de gonadotrofina sérica
V.5.2.9 Ensaio biológico de gonadotrofina coriônica
V.5.2.10 Ensaio biológico de gonadorelina
V.5.2.11 Ensaio biológico de menotrofina
V.5.2.12 Ensaio biológico de digital
V.5.2.13 Ensaio biológico de vasopressina
V.5.2.14 Ensaio biológico de lipressina
V.5.2.15 Ensaio biológico de felipressina
V.5.2.16 Ensaio biológico de somatotrofina
Método A: aumento do peso corporal
Método B: método da tíbia
V.5.2.17 Ensaio microbiológico de antibióticos
V.5.2.17.1 Ensaio microbiológico por difusão em ágar
V.5.2.17.2 Ensaio microbiológico por turbidimetria
VI PROCEDIMENTOS ESTATÍSTICOS APLICÁVEIS AOS ENSAIOS BIOLÓGICOS
VI.1 GLOSSÁRIO DE SÍMBOLOS
VI.2 FUNDAMENTOS
VI.3 VALORES ABERRANTES
VI.4 ENSAIOS DIRETOS
VI.5 ENSAIOS INDIRETOS QUANTITATIVOS
VI.5.1 Tipo de delineamento
VI.5.2 Análise de variância
VI.5.3 Testes de validade
VI.5.4 Estimativa da potência e limites de confiança
VI.6 MÉDIAS MÓVEIS
VI.7 ENSAIOS INDIRETOS “TUDO OU NADA”
VI.8 COMBINAÇÃO DE ESTIMATIVAS DE POTÊNCIA
VI.8.1 Potência média ponderada e limites de confiança
VI.9 TABELAS ESTATÍSTICAS
VI.10 EXEMPLOS DE ENSAIOS ESTATÍSTICOS
VI.10.1 Exemplo de ensaio direto
VI.10.2 Exemplo de ensaios indiretos quantitativos
VI.10.3 Exemplo de ensaio indireto “tudo ou nada”
VI.10.4 Exemplo de combinação de estimativas de potência
VII RADIOFÁRMACOS
VIII PRODUÇÃO DE DISCOS E METODOLOGIA PARA TESTE DE SENSIBILIDADE AOS ANTIBACTERIANOS
VIII.1 PRODUÇÃO DE DISCOS
VIII.2 CONTROLE DE DISCOS
IX RECIPIENTES E MATERIAIS EMPREGADOS NA SUA FABRICAÇÃO
IX.1 MATERIAS EMPREGADOS NA FABRICAÇÃO DE RECIPIENTES
IX.1.1 Material plástico
Métodos gerais de análise de materiais plásticos
Limpidez e grau de opalescência de soluções
Ensaio por combustão em atmosfera de oxigênio
IX.1.1.1 Materiais plásticos à base de cloreto de polivinila (PVC)
IX.1.1.2 Poliolefinas
IX.1.1.2.1 Polietileno de baixa densidade
IX.1.1.2.2 Polietileno de alta densidade
IX.1.1.2.3 Polipropileno
IX.1.1.2.4 Poliestireno
IX.1.1.2.5 Poliestireno opaco
IX.2 RECIPIENTES
IX.2.1 Recipiente de vidro
IX.2.2 Recipiente de material plástico
IX.2.2.1 Recipiente de material plástico para soluções injetáveis aquosas
IX.2.2.1.1Recipiente à base de cloreto de polivinila
IX.2.2.2 Recipiente de material plástico para sangue e produtos do sangue
IX.2.2.2.1 Recipiente à base de cloreto de polivinila para sangue e produtos do sangue, contendo ou não solução anticoagulante
X MÉTODOS DE PREPARAÇÃO
X.1 MÉTODOS DE ESTERILIZAÇÃO
X.1.1 Métodos físicos
X.1.1.1 Esterilização por calor
X.1.1.2 Esterilização por radiação
X.1.1.3 Esterilização por filtração
X.1.2 Método químico
X.1.2.1 Esterilização pelo óxido de etileno
X.2 INDICADORES BIOLÓGICOS
XI SUBSTÂNCIAS CORANTES
XII REAGENTES
XII.1 INDICADORES
XII.2 REAGENTES E SOLUÇÕES REAGENTES
XII.3 SOLUÇÕES VOLUMÉTRICAS
XII.4 TAMPÕES
XIII ANEXOS
XII.1 METODOLOGIA PARA O TESTE DE SENSIBILIDADEAOS ANTIBACTERIANOS (ANTIBIOGRAMA)
XIII.2 ANIMAIS DE LABORATÓRIO
XIII.2.1 Condições sanitárias
XIII.2.2 Ambiente
XIII.2.3 Nutrição
XIII.2.4 Genética
XIII.2.5 Ética
XII.3 NOMES, SÍMBOLOS E MASSAS ATÔMICAS
XII.4 UNIDADES DO SISTEMA INTERNACIONAL (SI) USADAS NA FARMACOPÉIA E EQUIVALÊNCIA COM OUTRAS UNIDADES
XII.5 MICRORGANISMOS EMPREGADOS EM TESTES E ENSAIOS TEXTOS QUE SUBSTITUEM OS PUBLICADOS, ANTERIORMENTE, NA PARTE II
GLICOSE
Dextrosum
C6H12O6 180,16 0627.01-1
C6H12O6.H2O 198,17
α -D-glicopiranose
Contém, no mínimo, 99% e, no máximo, 101,5% em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Cristais incolores ou pó cristalino branco, inodoro, de sabor doce.
Solubilidade. Facilmente solúvel em água e pouco solúvel em etanol a 96%.
Constantes físico-químicas
Poder rotatório específico (V.2.8): + 52,5º a + 53,5º (ver Ensaios de pureza).
IDENTIFICAÇÃO
A A 5 ml de solução quente de tartarato cúprico alcalino SR, adicionar algumas gotas de solução a 5% (p/V) da amostra. Forma-se precipitado vermelho de óxido cuproso.
B. Proceder conforme descrito no teste A de Identificação na monografia manitol, utilizando como referência glicose anidra padrão. A mancha principal, obtida no cromatograma da solução (1), é similar, em posição, cor e tamanho, à mancha obtida no cromatograma da solução (2).
ENSAIOS DE PUREZA
Cor da solução. Dissolver 12,5 g em água e completar o volume para 25 ml com o mesmo solvente. Esta solução não deverá é mais colorida que solução preparada pela mistura de 1 ml de cloreto cobaltoso SR, 3 ml de cloreto férrico SR e 2 ml de sulfato cúprico SR em água suficiente para 10 ml, diluindo-se, em seguida, 1,5 ml desta solução com água para obter 25 ml. Fazer a comparação sobre fundo branco em tubos de Nessler.
Acidez. Dissolver 5 g da amostra em 50 ml de água isenta de dióxido de carbono, adicionar fenolftaleína SI e titular com hidróxido de sódio 0,02 M SV até cor rósea. Deve ser necessário, no máximo, 0,3 ml para neutralização.
Poder rotatório específico (V.2.8). Determinar na substância previamente dessecada a 105 ºC. Preparar solução de 0,1 g/ml de hidróxido de amônio 0,012 M, deixar em repouso por 30 minutos e efetuar a leitura em tubo de 10 cm em polarímetro calibrado a 20 ºC. A leitura deve estar entre + 52,5o e + 53,2o a 20 oC.
Água (V.2.20.1). De 7 a 9,5%, para glicose monoidratada, e, no máximo, 1% para glicose anidra. Determinar em 0,5 g.
Arsênio (V.3.2.5). Determinar em 1 g de amostra. No máximo 0,0001% (1 ppm).
Cloretos (V.3.2.1). Determinar em 2 g de amostra. No máximo 0,018% (180 ppm).
Sulfatos (V.3.2.2). Determinar em 2 g de amostra em comparação a 0,5 ml de ácido sulfúrico 0,01 M. Máximo 0,025% (250 ppm) .
Metais pesados (V.3.2.3). Proceder ao ensaio em solução preparada pela dissolução de 4 g em água e completando o volume para 25 ml.No máximo 0,0005% (5 ppm) .
Dextrinas e açúcares menos solúveis. Dissolver 1 g de amostra pulverizada em 30 ml de etanol 90% sob aquecimento e agitação em balão dotado de coluna de refluxo. A solução deve permanecer límpida depois de esfriar.
Amido solúvel e sulfitos. Dissolver 1 g em 10 ml de água e adicionar uma gota de iodo 0,1 M SV. A solução cora-se de amarelo, não devendo aparecer cor azul.
TESTES DE SEGURANÇA BIOLÓGICA
Contagem microbiana (V.5.1.6). Cumpre o teste.
Pesquisa e identificação de patógenos (V.5.1.7). Cumpre o teste.
Pirogênios (V.5.1.2). A glicose para preparação de soluções parenterais de grande volume deve ser testada quanto à ausência de pirogênios.
Injetar 10 ml/kg de uma solução em água para injeção contendo 50 mg/ml de glicose.
DOSEAMENTO
Pesar exatamente cerca de 0,1 g e dissolver em 50 ml de água, em frasco provido de tampa esmerilhada. Adicionar 25 ml de iodo 0,05 M SV e 10 ml de carbonato de sódio SR. Homogeneizar e deixar em repouso por 20 minutos protegidos da luz. Adicionar 15 ml de ácido clorídrico diluído SR e titular o excesso de iodo com tiossulfato de sódio 0,1 M SV, usando amido SI como indicador. Efetuar ensaio em branco. Cada ml da solução de iodo 0,05 M SV consumido corresponde a 9,008 mg de C6H12O6 e a 9,908 mg de C6H12O6.H2O.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da luz, à temperatura ambiente.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Energético e excipiente.
76
AMOXICILINA TRIIDRATADA
Amoxicillinum trihydricum
Ácido[2S-[2α,5α,6β(S∗)]]-6-[[amino(4-hidroxifenil)acetil]amino]-3,3-dimetil-7-oxo-4-tia-1-azabiciclo[3.2.0]heptano-2-carboxílico triidratado.
Apresenta potência de, no mínimo, 900 μg e, no máximo, 1050 μg de amoxicilina por miligrama, em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Pó cristalino branco ou quase branco.
Solubilidade. Pouco solúvel em água, etanol e metanol. Insolúvel em benzeno, hexano, acetato de etila, clorofórmio, éter e acetonitrila.
Dissolve em soluções de hidróxidos alcalinos.
Constantes físico-químicas
Poder rotatório específico (V.2.8): + 290o a + 315o, determinado em solução a 0,2% (p/V) em água isenta de dióxido de carbono, calculado em relação à substância anidra.
IDENTIFICAÇÃO
O teste de identificação A pode ser omitido se forem realizados os testes B e C. Os testes de identificação B e C podem ser omitidos se for realizado o teste A.
A. O espectro de absorção no infravermelho (V.2.14-4) da amostra dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de amoxicilina triidratada padrão, preparado de maneira idêntica.
B. O espectro de absorção no ultravioleta (V.2.14-3), na faixa de 200 a 400 nm, de uma solução a 0,02% (p/V), em etanol, exibe máximos em 230 nm e em 274 nm, idênticos aos observados no espectro de solução similar do padrão.
C. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel G 60, como suporte, e mistura de metanol-clorofórmio-água-acetona (9:8:3:1), como fase móvel. Aplicar, separadamente, à placa, 5 μ l de cada uma das soluções descritas a seguir:
Solução (1): solução a 0,4% (p/V) da amostra em ácido clorídrico 0,1 M, que deve ser usada, no máximo, 10 minutos após de sua preparação.
Solução (2): solução a 0,4% (p/V) do padrão em ácido clorídrico 0,1 M, que deve ser usada, no máximo, 10 minutos após de sua preparação.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e nebulizar com solução contendo 0,3% (p/V) de ninidrina em álcool.
Aquecer em estufa a 110 ºC por 15 minutos. A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidadeàquela obtida com a solução (2).
ENSAIOS DE PUREZA
pH (V.2.19). 3,5 a 6,0. Determinar em solução aquosa a 0,2% (p/V).
Água (V.2.20.1). 11,5 a 14,5%. Determinar em 0,3 g de amostra.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 1%.
Cristalinidade. Suspender algumas partículas da amostra em óleo mineral, transferir para uma lâmina de vidro e examinar por meio de microscópio dotado de luz polarizada. As partículas exibem birrefringência, que se extingue ao movimentar a amostra por meio de ajuste micrométrico.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Dissolver padrão e amostra em água estéril, de modo a obter solução na concentração de, aproximadamente, 1 mg/ml. Diluir solução padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Dissolver e diluir padrão e amostra de amoxicilina, em água, até concentração, aproximada, de 1 mg/ml.
Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI, e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a amostra. Realizar prova em branco da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções, adicionadas de 10 ml de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI, e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
Em que
P= potência da amostra (μg/mg);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
Pó = potência do padrão (μg/mg);
Pp = peso do padrão (mg);
Pa = peso da amostra (mg).
EMBALAGEM E ARMAZENAMENTO
Em recipientes perfeitamente fechados, protegidos do ar e da luz, a temperatura inferior a 30 ºC.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antibiótico
76.1
AMOXICILINA TRIIDRATADA CÁPSULAS
Cápsulas de amoxicilina triidratada são constituídas de amoxicilina triidratada com ou sem, um ou mais, agentes lubrificantes, diluentes e secantes adequados, incluídos em cápsula de gelatina. A potência é de, no mínimo, 90,0% e, no máximo, 120,0% do valor declarado de amoxicilina.
IDENTIFICAÇÃO
A. Proceder conforme descrito no teste B de Identificação na monografia
Amoxicilina triidratada.
B. Proceder conforme descrito no teste C de Identificação na monografia
Amoxicilina triidratada.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: água, 900 ml
Aparelhagem: cesta, 100 rpm
Tempo: 90 minutos
Procedimento: após o teste, retirar alíquota do meio de dissolução, filtrar e diluir em água, até concentração adequada. Medir as absorvâncias das soluções em 272 nm (V.2.14-3), utilizando o mesmo solvente para zerar o aparelho. Calcular a quantidade de C16H19N3O5S dissolvida no meio, comparando as leituras obtidas com a da solução padrão na concentração de 0,01% (p/V) preparada no mesmo solvente.
Tolerância: não menos que 80% da quantidade declarada de C16H19N3O5S se dissolvem em 90 minutos.
ENSAIOS DE PUREZA
Água (V.2.20.1). No máximo 14,5%. Determinar em 0,3 g da amostra.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir, usando amostragem de 20 cápsulas.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Dissolver o padrão de amoxicilina em água estéril, de modo a obter solução na concentração de, aproximadamente, 1 mg/ml. Pesar 20 cápsulas, remover o conteúdo e pesá-las novamente. Homogeneizar o conteúdo e transferir quantidade, exatamente pesada, para frasco volumétrico. Diluir com tampão fosfato de potássio 0,1 M, estéril, pH 8 (Solução 2) para obter solução de trabalho na concentração da solução padrão. Agitar de 3 a 5 minutos.
Diluir solução padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Dissolver o padrão de amoxicilina emágua, até concentração, aproximada, de 1 mg/ml. Remover o conteúdo das cápsula e pesar exatamente. Misturar os conteúdos. Transferir quantidade representativa da amostra, exatamente pesada, para frasco volumétrico e diluir com água de modo a obter concentração idêntica à do padrão. Agitar de 3 a 5 minutos. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar prova em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções, adicionadas de 10 ml de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
Em que
P = potência da amostra (mg/cápsula);
Vba = volume do titulante gasto na titulação da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vp = volume do titulante gasto na titulação do padrão (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
S =concentração do padrão (mg/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, entre 15 e 25 ºC.
ROTULAGEM
Observar a legislação vigente.
76.2
AMOXICILINA TRIIDRATADA PÓ PARA SUSPENSÃO ORAL
Amoxicilina triidratada pó para suspensão oral é mistura de um ou mais agentes adequados para suspensão, contendo ou não corantes, aromatizantes, conservantes, tampões, adoçantes e estabilizantes. A potência é de, no mínimo, 90,0% e, no máximo, 120,0% do valor declarado de amoxicilina declarada.
IDENTIFICAÇÃO
Proceder conforme descrito no teste C de Identificação na monografia Amoxicilina triidratada.
CARACTERÍSTICAS
pH (V.2.19). 5,0 a 7,5. Determinar na suspensão reconstituída, conforme indicação do produtor.
Determinação de peso (V.1.1). Cumpre o teste.
ENSAIO DE PUREZA
Água (V.2.20.1). No máximo 3%. Determinar em 0,3 g da amostra.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos
(V.5.2.17). Dissolver o padrão de amoxicilina em água
estéril, de modo a obter solução na concentração de, aproximadamente,
1 mg/ml. Reconstituir o conteúdo conforme indicado pelo
produtor. Transferir quantidade exatamente medida, para frasco volumétrico
e diluir com tampão fosfato de potássio 0,1 M, estéril, pH
8,0 (Solução 2) para obter solução de trabalho na concentração da
solução padrão. Diluir solução padrão e amostra, em tampão fosfato
de potássio 0,1 M, estéril, pH 8,0 (solução 2), às concentrações
empregadas na curva padrão.
B. Por método iodométrico. Dissolver o padrão de amoxicilina em água, até concentração, aproximada, de 1mg/ml. Reconstituir a amostra conforme indicado pelo produtor. Transferir quantidade representativa da amostra para frasco volumétrico e diluir com água de modo a obter concentração idêntica à do padrão. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar prova em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções, adicionadas de 10 ml de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV. Em que
P = potência da amostra (mg/frasco);
Vba = volume do titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vp = Volume de titulante gasto na titulação do padrão (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
S = concentração do padrão (mg/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes perfeitamente fechados, entre 15 e 25 ºC.
ROTULAGEM
Observar a legislação vigente.
77
AMPICILINA
Ampicillinum
C16H19N304S 349,41 0061.01-8
Ácido [2S-[2α,5α,6β(S∗)]]-6-[(aminofenacetil)amino]-3,3-dimetil-7- oxo-4-tia-1-azabiciclo [3.2.0]heptano-2-carboxílico.
Apresenta potência de, no mínimo, 900 μg e, no máximo, 1 050 μg de ampicilina por miligrama, em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Pó cristalino branco a levemente amarelado.
Solubilidade. Levemente solúvel em água e em metanol; praticamente insolúvel em acetona, em clorofórmio, em etanol absoluto, em éter etílico; insolúvel em benzeno e em tetracloreto de carbono. Dissolve em soluções ácidas e alcalinas diluídas.
Constantes físico-químicas
Faixa de fusão (V.2.2): 199 ºC a 202 ºC.
Poder rotatório específico (V.2.8): + 280º a + 305º, determinado em solução a 0,25% (p/V), calculado em relação à substância anidra.
IDENTIFICAÇÃO
O teste de identificação A pode ser omitido se forem realizados os testes B e C. Os testes de identificação B e C podem ser omitidos se for realizado o teste A.
A. O espectro de absorção no infravermelho (V.2.14-4) da amostra dispersa em brometo de potássio apresenta máximos de absorção nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro da ampicilina padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando como suporte sílica-gel H, e como fase móvel acetona-acetato de amônio 15,4% (p/V) (90:10), pH 5,0, ajustado comácido acético glacial. Aplicar, separadamente, à placa 2 μl de cada uma das soluções descritas a seguir, recentemente preparadas.
Solução (1): solução a 0,25% (p/V) da amostra em solução de bicarbonato de sódio a 4,2%.
Solução (2): solução a 0,25% (p/V) do padrão em solução de bicarbonato de sódio a 4,2%.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e expor a vapores de iodo até o aparecimento das manchas. Examinar sob luz visível. A mancha principal obtida no cromatograma com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (2).
C. Transferir cerca de 2 mg da amostra para tubo de ensaio. Umedecer com 0,05 ml de água e adicionar 2 ml da mistura de 2 ml de solução de formaldeído com 100 ml de ácido sulfúrico. Agitar o tubo e observar a cor. Deve-se apresentar quase incolor. Imergir o tubo em banho-maria fervente durante um minuto. Desenvolve-se coloração amarela escura.
ENSAIOS DE PUREZA
pH (V.2.19). 3,5 a 6,0. Determinar em solução aquosa a 1% (p/V).
Perda por dessecação. Proceder conforme descrito em Dessecação de substâncias antibióticas (V.5.2.17-4-Método 1). No máximo 2%.
Cinzas sulfatadas (V.2.10). Determinar em 1 g de amostra. No máximo 0,5%.
Cristalinidade. Suspender algumas partículas da amostra em óleo mineral, transferir para uma lâmina de vidro e examinar por meio de microscópio dotado de luz polarizada. As partículas exibem birrefringência, que se extingue ao movimentar a amostra por meio de ajuste micrométrico.
N,N-Dimetilanilina. No máximo 0,02% (200 ppm). Proceder conforme descrito em Cromatografia a gás (V.2.17.5) utilizando cromatógrafo provido de detector de ionização de chama; coluna de vidro (2 m x 2 mm) empacotada com suporte de diatomáceas silanizado, impregnado com 3% (p/p) de fenilmetilsilicone (50% fenil), mantida a 120 ºC; injetor e detector a 150 ºC, gás de arraste nitrogênio para cromatografia, fluxo de 30 ml/minuto.
Solução de padrão interno (solução A): solução de naftaleno a 0,005% (p/V) em ciclohexano.
Solução de dimetilanilina padrão: dissolver 50 mg de N,N-dimetilanilina
em mistura de 2 ml de ácido clorídrico em 20 ml de água
sob agitação, completar o volume a 50 ml com água e agitar. Transferir
5 ml para balão volumétrico de 250 ml, completar o volume com
água e agitar. Transferir 1 ml para tubo de ensaio, adicionar 5 ml de
hidróxido de sódio M, 1 ml da solução A, agitar vigorosamente por 1
minuto, centrifugar, se necessário, e usar o sobrenadante.
Solução amostra: dissolver 1 g da amostra em 5 ml de hidróxido de sódio 1 M, adicionar 1 ml da amostra A, agitar vigorosamente por 1 minuto, centrifugar, se necessário, e usar o sobrenadante.
Procedimento: injetar, separadamente, 1 μl das soluções padrão e amostra, registrar os cromatogramas e medir as áreas dos picos principais.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Dissolver padrão e amostra em água estéril de modo a obter solução na concentração de 0,1 mg/ml. Diluir solução padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Dissolver e diluir padrão e amostra de
ampicilina sódica em água, até concentração de, aproximadamente,
1,25 mg/ml. Transferir 2 ml da solução padrão para erlenmeyer com
tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar
e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico
1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em
repouso, por 15 minutos, ao abrigo da luz. Titular com solução de
tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3
gotas de solução de amido SI e prosseguir com a titulação até o
desaparecimento da cor azul. Proceder ao mesmo ensaio com a solução
amostra. Realizar prova em branco, da amostra e do padrão, por
meio da titulação de 2 ml de ambas soluções, adicionadas de 10 ml da
solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo
ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato
de sódio 0,01 M SV.
Em que
P = potência da amostra (μg/mg);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
Pó = potência do padrão (μg/mg);
Pp = peso do padrão (mg);
Pa = peso da amostra (mg).
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 30ºC.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antibiótico.
77.1
AMPICILINA CÁPSULAS
Cápsulas de ampicilina são constituídas de ampicilina triidratada com ou sem, agentes tampões, lubrificantes, diluentes, aglutinantes, óleos vegetais, corantes e aromatizantes adequados, incluídos em cápsulas de gelatina. A potência é de, no mínimo, 90,0% e, no máximo, 120,0% do valor declarado de ampicilina.
IDENTIFICAÇÃO
A. Proceder conforme descrito no teste B de Identificação na monografia
Ampicilina.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: água, 900 ml
Aparelhagem: cesto, 100 rpm
Tempo: 45 minutos
Procedimento: após o teste, retirar alíquota do meio de dissolução, filtrar e diluir em tampão sulfato de cobre, até concentração adequada.
Transferir alíquota de 10 ml para tubo de ensaio com tampa,
submeter a aquecimento em banho-maria a 75 oC por 30 minutos e
resfriar rapidamente. Medir as absorvâncias das soluções a 320 nm
(V.2.14-3) utilizando a solução amostra sem aquecimento para ajuste
do zero do aparelho. Calcular a quantidade de C16H19N3O4S dissolvida
no meio, comparando as leituras obtidas com a solução padrão
na concentração de 0,0022% (p/V) preparada nas mesmas condições.
Tolerância: não menos que 75% da quantidade declarada de ampicilina dissolvem-se em 45 minutos.
ENSAIOS DE PUREZA
Perda por dessecação. Proceder conforme descrito em Dessecação de substâncias antibióticas (V.5.2.17-4-Método 1). No máximo 4%.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir, usando amostragem de, pelo menos, 20 cápsulas.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Dissolver o padrão de ampicilina em água estéril, de modo a obter solução na concentração de, aproximadamente, 0,1 mg/ml. Remover o conteúdo das cápsulas e pesa-las novamente.
Homogenizar os conteúdos e transferir uma quantidade, exatamente pesada, para frasco volumétrico e diluir com tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) para obter solução de trabalho na concentração da solução padrão. Agitar de 3 a 5 minutos.
Diluir solução padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Dissolver o padrão de ampicilina emágua, até concentração, aproximada, de 1,25 mg/ml. Remover o conteúdo das cápsulas e pesar exatamente. Misturar os conteúdos. Transferir quantidade representativa da amostra, exatamente pesada, para frasco volumétrico e diluir com água de modo a obter concentração idêntica à do padrão. Agitar de 3 a 5 minutos. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar prova em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções, adicionadas de 10 ml de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. . Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
Em que
P = potência da amostra (mg/cápsula);
Vba = volume de titulante gasto na titulação do branco da amostra(ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (mg/ml)
F = fator de diluição da amostra
EMBALAGEM E ARMAZENAMENTO
Em recipientes perfeitamente fechados, entre 15 e 25 ºC.
ROTULAGEM
Observar a legislação vigente.
XII.4 TAMPÕES
Tampão de sulfato de cobre
Preparação: no momento de uso, misturar 15 ml de solução B em 985 ml de solução A.
Solução A: dissolver 15,22 g de fosfato (dibásico) de sódio anidro em
quantidade suficiente de água. Adicionar 9,75 g de ácido cítrico
monoidratado e completar o volume para 1000 ml com água. Acertar
a pH 5,2 com auxílio de hidróxido de sódio ou ácido cítrico.
Solução B: dissolver 0,313 g de sulfato de cobre pentaidratado em
100 ml de água.
77.2
AMPICILINA COMPRIMIDOS
Comprimidos de ampicilina são constituídos de ampicilina com um ou mais agentes diluentes e lubrificantes adequados. A potência é de, no mínimo, 90,0% e, no máximo, 120,0% do valor declarado de ampicilina.
IDENTIFICAÇÃO
Proceder conforme descrito no teste B de Identificação na monografia
Ampicilina.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). No máximo 15 minutos.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: água, 900 ml
Aparelhagem: cesta, 100 rpm
Tempo: 45 minutos
Procedimento: após o teste, retirar alíquotas do meio de dissolução, filtrar e diluir, em tampão de sulfato de cobre, até concentração adequada. Prosseguir conforme descrito no Teste de dissolução em Ampicilina cápsulas.
Tolerância: não menos que 75% da quantidade declarada de ampicilina se dissolvem em 45 minutos.
ENSAIOS DE PUREZA
Perda por dessecação. Proceder conforme descrito em Dessecação de substâncias antibióticas (V.5.2.17-4-Método 1). No máximo 4%.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir, usando amostragem de, pelo menos, 10 comprimidos.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Dissolver o padrão de ampicilina em água estéril, de modo a obter solução na concentração de, aproximadamente, 0,1 mg/ml. Pesar e pulverizar os comprimidos. Transferir quantidade representativa da amostra, exatamente pesada, para frasco volumétrico e diluir com tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) para obter solução de trabalho na concentração da solução padrão. Agitar de 3 a 5 minutos. Diluir solução padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Dissolver o padrão de ampicilina emágua, até concentração, aproximada, de 1,25 mg/ml. Pesar e pulverizar os comprimidos. Transferir quantidade representativa da amostra, exatamente pesada, para balão volumétrico e diluir com água de modo a obter concentração idêntica à do padrão. Agitar de 3 a 5 minutos. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução d tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar prova em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções, adicionadas de 10 ml de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
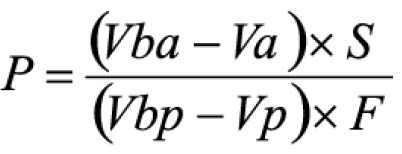
Em que
P = Potência da amostra (mg/comprimido);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (mg/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes perfeitamente fechados, protegidos da umidade e a temperatura inferior a 25 ºC.
ROTULAGEM
Observar a legislação vigente.
_________________________________________________
XII.4 TAMPÕES
Tampão de sulfato de cobre
Preparação: no momento de uso, misturar 15 ml de solução B em 985 ml de solução A.
Solução A: dissolver 15,22 g de fosfato (dibásico) de sódio anidro em
quantidade suficiente de água. Adicionar 9,75 g de ácido cítrico
monoidratado e completar o volume para 1000 ml com água. Acertar
a pH 5,2 com auxílio de hidróxido de sódio ou ácido cítrico.
Solução B: dissolver 0,313 g de sulfato de cobre pentaidratado em
100 ml de água.
77.3
AMPICILINA PÓ PARA SUSPENSÃO ORAL
Ampicilina pó para suspensão oral é mistura de ampicilina com um ou mais agentes corantes, aromatizantes, tampões, adoçantes e conservantes.
A potênica é de, no mínimo, 90,0% e, no máximo, 120,0% do valor declarado de ampicilina.
IDENTIFICAÇÃO
Proceder conforme descrito no teste B de Identificação na monografia
Ampicilina.
CARACTERÍSTICAS
pH (V.2.19). 5,0 a 7,5. Determinar na suspensão reconstituída, conforme indicação do produtor.
Determinação do peso (V.1.1). Cumpre o teste.
Determinação do volume (V.1.2). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
ENSAIOS DE PUREZA
Água (V.2.20.1). No máximo 2,5%, em quantidade contendo o equivalente a 100 mg/ml de ampicilina após a reconstituição.
TESTES DE SEGURANÇA BIOLÓGICA
Contagem de microrganismos viáveis (V.5.1.6.1). Bactérias aeróbicas totais: no máximo 1 000 UFC/ml. Fungos e leveduras: no máximo 100 UFC/ml.
Pesquisa e identificação de patógenos (V.5.1.7). Cumpre o teste.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Dissolver o padrão de ampicilina em água estéril, de modo a obter solução na concentração de, aproximadamente, 0,1 mg/ml. Reconstituir o conteúdo conforme indicado pelo produtor.
Transferir quantidade representativa da amostra para frasco volumétrico e diluir com tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) para obter solução de trabalho na concentração da solução padrão. Agitar de 3 a 5 minutos. Diluir solução padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Dissolver o padrão de ampicilina emágua, até concentração, aproximada, de 1,25 mg/ml. Reconstituir o conteúdo conforme indicado pelo produtor. Transferir quantidade representativa da amostra para balão volumétrico e diluir com água de modo a obter concentração idêntica à do padrão. Agitar de 3 a 5 minutos. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio 1 M.
Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácidoclorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 mnutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar provas em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções, adicionadas de 10 ml de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
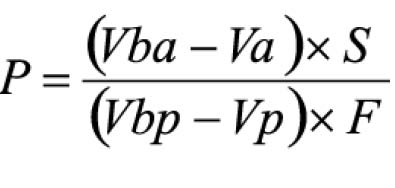
Em que
P = potência da amostra (mg/frasco);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (mg/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da umidade e em temperatura inferior a 25 ºC.
ROTULAGEM
Observar a legislação vigente.
78
AMPICILINA SÓDICA
Ampicillinum natricum
C16H18NaN304S 371,39 0061.04-2
Sal monossódico do ácido [2S-[2α,5α,6β(S∗)]]-6-[(aminofenacetil) amino]-3,3-dimetil-7-oxo-4-tia-1-azabiciclo[3.2.0]heptano-2-carboxílico.
Apresenta potência de, no mínimo, 845 μg e, no máximo, 988 μg de ampicilina por miligrama, calculado em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Pó cristalino branco, higroscópico.
Solubilidade. Muito solúvel em água, solúvel em acetona, pouco solúvel em clorofórmio, praticamente insolúvel em éter.
Constantes físico-químicas
Faixa de fusão (V.2.2): 203 ºC a 206 ºC.
Poder rotatório específico (V.2.8): + 258º a + 287º, determinado em solução a 0,25% (p/V) tendo como solvente solução de biftalato de potássio a 0,4% (p/V), calculado em relação à substância anidra.
IDENTIFICAÇÃO
O teste de identificação A pode ser omitido se forem realizados os testes B e C. Os testes de identificação B e C podem ser omitidos se for realizado o teste A
A. Dissolver 250 mg em 5 ml de água, adicionar 0,5 ml de ácido acético 2 M, agitar e deixar em repouso por 10 minutos em banho de gelo. Filtrar através de filtro de vidro sinterizado, sob pressão reduzida, lavar com 2 a 3 ml de mistura de 9 partes de acetona e 1 parte de água e secar a 60 ºC por 30 minutos. O espectro de absorção no infravermelho (V.2.14-4) da amostra dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de ampicilina sódica padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando como suporte sílica-gel GF-254, e como fase móvel mistura de acetona-acetato de amônio a 15,4% (p/V) (90: 10), com pH 5,0, ajustado com ácido acético glacial. Aplicar, separadamente,à placa 2 μl de cada uma das soluções, recentemente preparadas, descritas a seguir.
Solução (1): solução a 0,5% (p/V) da amostra em solução de bicarbonato de sódio a 4,2% (p/V).
Solução (2): solução a 0,5% (p/V) do padrão em solução de bicarbonato de sódio a 4,2% (p/V).
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e nebulizar com solução alcoólica de ninidrina a 0,3% (p/V), aquecer em estufa de calor seco, a 90 ºC, durante 15 minutos. Examinar sob luz visível. A mancha principal obtida no cromatograma com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (2).
C. Transferir cerca de 2 mg da amostra para tubo de ensaio. Umedecer com 0.05 ml de água e adicionar 2 ml da mistura de 2 ml de solução de formaldeído com 100 ml de ácido sulfúrico. Agitar o tubo e observar a cor. Deve-se apresentar quase incolor. Imergir o tubo em banho-maria durante um minuto. Desenvolve-se coloração marromavermelhada.
D. Atende ao teste de Identificação para o íon sódio (V.3.1).
ENSAIOS DE PUREZA
pH (V.2.19). 8,0 a 10,0. Determinar em solução aquosa a 1% (p/V).
Água (V.2.20.1). Não mais que 2,0%.
Cristalinidade. Suspender algumas partículas da amostra em óleo mineral, transferir para uma lâmina de vidro e examinar por meio de microscópio dotado de luz polarizada. As partículas exibem birrefringência, que se extingue ao movimentar a amostra por meio de ajuste micrométrico.
N,N-Dimetilanilina. No máximo 0,02% (200 ppm). Proceder conforme descrito em Ensaios de Pureza na monografia Ampicilina.
Diclorometano. Não mais que 0,2% (p/p) quando determinado por cromatografia a gás (V.2.17.5) utilizando cromatógrafo provido de detector de ionização de chama: coluna de vidro (105 m x 4 mm) empacotada com suporte de diatomáceas silanizado (partículas de até 120 μm), lavado com ácido, revestido com 10% (p/p) de polietilenoglicol 1 000, mantida a 60 ºC; injetor a 100 ºC; detector a 150ºC, gás de arraste nitrogênio para cromatografia, fluxo de 40 ml/mimuto.
Solução de padrão de diclorometano: transferir 1 ml de solução aquosa de diclorometano 0,2% (V/V) para balão volumétrico de 100 ml.
Acrescentar 1 ml da solução aquosa de 1,2-diclorometano 0,2% (V/V)
(Padrão interno), completar o volume com água e agitar.
Solução amostra: dissolver 10 g da amostra em água e transferir para balão volumétrico de 100 ml. Adicionar 1,0 ml de solução aquosa a 0,2% (V/V) de 1,2-diclorometano (Padrão interno), completar o volume com água e agitar.
Procedimento: injetar, separadamente, 1 μl das soluções padrão e amostra, registar os cromatogramas e calcular a porcentagem (p/p) de diclorometano, considerando como 1,325 g/ml o valor da densidade a 20 ºC.
TESTES DE SEGURANÇA BIOLÓGICA
Ampicilina sódica destinada à preparação parenteral deve cumprir com os seguintes testes adicionais.
Esterilidade. (V.5.1.1-5). Cumpre o teste. Empregar método de filtração por membranas. Dissolver 6 g da amostra em 800 ml de Fluido II contendo quantidade suficiente de penicilinase estéril para inativar a ampicilina, agitar até total solubilização e proceder como descrito.
Pirogênio. (V.5.1.2). Cumpre o teste. Injetar, 1 ml/kg, empregando solução de ampicilina sódica 20 mg/ml, em água.
Endotoxinas bacterianas. (V.5.1.9). No máximo 0,15 UE/mg de ampicilina.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos
(V.5.2.17). Dissolver padrão e amostra em água estéril de modo a obter solução na concentração de 0,1mg/ml. Diluir solução padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Dissolver e diluir padrão e amostra de ampicilina sódica em água, até concentração de, aproximadamente, 1,25 mg/ml. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder ao mesmo ensaio com a solução amostra. Realizar prova em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções, adicionadas de 10 ml da solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M.
Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
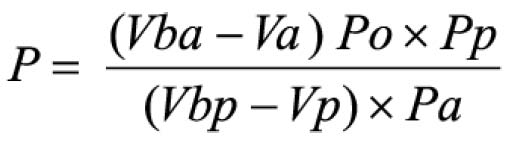
Em que
P = potência da amostra (μg/mg);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
Pó = potência do padrão (μg/mg);
Pp = peso do padrão (mg);
Pa = peso da amostra (mg).
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 30ºC. Sendo destinado à produção de formas farmacêuticas injetáveis, deverá ser embalao em recipientes estéreis.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antibiótico.
78.1
AMPICILINA SÓDICA PÓ PARA SOLUÇÃO INJETÁVEL
A potência é de, no mínimo, 90,0% e, no máximo, 115,0% do valor declarado de ampicilina.
IDENTIFICAÇÃO
Proceder conforme descrito no teste B de Identificação na monografia
Ampicilina sódica.
CARACTERÍSTICAS
pH (V.2.19). 8,0 a 10,0. Determinar em solução aquosa a 1% (p/V).
Determinação do peso (V.1.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
ENSAIOS DE PUREZA
Água (V.2.20.1). Não mais que 2,0%.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1-5). Cumpre o teste. Empregar método de filtração por membranas. Dissolver 6 g da amostra em 800 ml de Fluido II contendo quantidade suficiente de penicilinase estéril para inativar a ampicilina, agitar até total solubilização e proceder como descrito.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 1,0 ml/kg, empregando solução de ampicilina sódica 20 mg/ml, em água.
Endotoxinas bacterianas (V.5.1.9). No máximo 0,15 UE/mg de ampicilina.
DETERMINAÇÃO DA POTÊNCIA
Para determinação da potência do pó para solução injetável de ampicilina, empregar um dos métodos descritos a seguir, utilizando amostragem mínima de 10 frascos.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Dissolver padrão e amostra em água estéril de modo a obter solução na concentração de 0,1 mg/ml. Diluir solução padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Dissolver e diluir padrão e amostra reconstituída em água, até concentração de, aproximadamente, 1,25 mg/ml. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder ao mesmo ensaio com a solução amostra. Realizar provas em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções, adicionadas de 10 ml da solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M.
Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
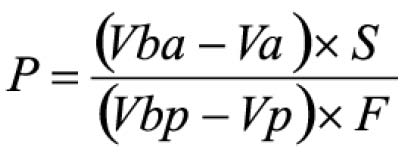
Em que
P = potência da amostra (mg/frasco-ampola);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (mg/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 25ºC.
ROTULAGEM
Observar a legislação vigente.
79
AMPICILINA TRIIDRATADA
Ampicillinum trihydricum
Ácido [2S-[2α,5α,6β(S∗)]]-6-[(aminofenacetil)amino]-3,3-dimetil-7- oxo-4-tia-1-azabiciclo[3.2.0]heptano-2-carboxílico triidratado.
Apresenta potência de, no mínimo, 900 μg e, no máximo, 1 050 μg de ampicilina por miligrama, em relação à substância anidra.
DESCRIÇÃO
Caracteres físicos. Pó cristalino branco.
Solubilidade. Pouco solúvel em água, praticamente insolúvel em clorofórmio, etanol, éter etílico e em óleos fixos. Dissolve-se em soluções de hidróxidos alcalinos.
IDENTIFICAÇÃO
O teste de identificação A pode ser omitido se forem realizados os testes B e C. Os testes de identificação B e C podem ser omitidos se for realizado o teste A.
A. O espectro de absorção no infravermelho (V.2.14-4) da amostra dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de ampicilina triidratada padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Cromatografia em camada delgada
(V.2.17.1), utilizando como suporte sílica-gel H, e como fase móvel mistura de acetona-acetato de amônio a 15,4% (p/V) (10:90), pH 5,0, ajustado com ácido acético glacial. Aplicar, separadamente, à placa, 1μl de cada uma das seguintes soluções.
Solução (1): solução a 0,25% (p/V) da amostra em solução de bicarbonato de sódio a 4,2% (p/V).
Solução (2): solução a 0,25% (p/V) do padrão em solução de bicarbonato de sódio a 4,2% (p/V).
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e expor a vapores de iodo até o aparecimento das manchas. Examinar sob luz visível. A mancha principal obtida no cromatograma com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (2).
C. Transferir cerca de 2 mg da amostra para tubo de ensaio. Umedecer com 0,05 ml de água e adicionar 2 ml da mistura de 2 ml de solução de formaldeido com 100 ml de ácido sulfúrico. Agitar o tubo e observar a cor. Deve-se apresentar quase incolor. Imergir o tubo em banho-maria fervente durante um minuto. Desenvolve-se coloração amarela escura.
ENSAIOS DE PUREZA
pH (V.2.19). 3,5 a 6,0. Determinar em solução aquosa a 1% (p/V).
Perda por dessecação. Proceder conforme descrito em Dessecação de substâncias antibióticas (V.5.2.17-4-Método 1). De 12,0 a 15,0%.
Cristalinidade. Suspender algumas partículas da amostra em óleo mineral, transferir para uma lâmina de vidro e examinar por meio de microscópio dotado de luz polarizada. As partículas exibem birrefringência, que se extingue ao movimentar a amostra por meio de ajuste micrométrico.
TESTES DE SEGURANÇA BIOLÓGICA
Ampicilina triidratada destinada a preparação parenteral deve cumprir com os seguintes testes adicionais.
Esterilidade (V.5.1.1-5). Cumpre o teste. Empregar método de filtração por membranas. Dissolver 6 g da amostra em 800 ml de Fluido II contendo quantidade suficiente de penicilinase estéril para inativar a ampicilina, agitar até total solubilização e proceder conforme descrito.
Pirogênio (V.5.1.2). Cumpre o teste. Injetar 1 ml/kg, empregando solução de ampicilina na concentração de 20 mg/ml, em solução de hidróxido de sódio 0,05 M.
Endotoxinas bacterianas (V.5.1.9). No máximo 0,15 UE/mg de ampicilina.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Dissolver padrão e amostra em água estéril de modo a obter solução na concentração de, aproximadamente, 0,1 mg/ml. Diluir soluções padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Dissolver e diluir padrão e amostra de ampicilina triidratada, em água, até concentração de, aproximadamente, 1,25 mg/ml. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml deácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder ao mesmo ensaio com a solução amostra. Realizar prova em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas soluções, adicionadas de 10 ml de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
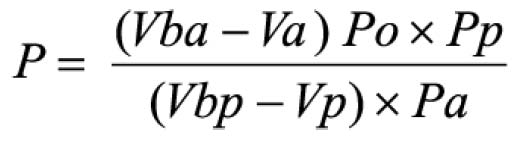
Em que
P = Potência da amostra (μg/mg);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
Pó = potência do padrão (μg/mg);
Pp = peso do padrão (mg);
Pa = peso da amostra (mg).
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 30ºC. Sendo destinado a produção de formas farmacêuticas injetáveis, deverá ser embalado em recipientes estéreis.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antibiótico.
79.1
AMPICILINA TRIIDRATADA CÁPSULAS
Ampicilina triidratada cápsulas são compostas de ampicilina triidratada com ou sem, um ou mais, agentes tampões, lubrificantes, diluentes, aglutinantes, óleos vegetais, corantes e aromatizantes adequados, incluídos em cápsulas de gelatina. A potência é de, no mínimo, 90,0% e, no máximo, 120,0% do valor declarado de ampicilina.
IDENTIFICAÇÃO
A. Proceder conforme descrito no teste B de Identificação na monografia
Ampicilina triidratada.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Teste de desintegração (V.1.4.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: água, 900 ml
Aparelhagem: cesto, 100 rpm
Tempo: 45 minutos
Procedimento: após o teste, retirar alíquota do meio de dissolução, filtrar e diluir em solução tampão de sulfato de cobre, até concentração adequada. Transferir alíquota de 10 ml para tubo de ensaio com tampa, submeter a aquecimento em banho-maria a 75 oC por 30 minutos e resfriar rapidamente. Medir a absorvância da solução em 320 nm (V.2.14-3) utilizando a solução amostra sem aquecimento para zerar o aparelho. Calcular a quantidade de C16H19N3O4S dissolvida no meio, comparando as leituras obtidas com a da solução padrão de referência na concentração de 0,0022% (p/V) preparada nas mesmas condições.
Tolerância: não menos que 75% da quantidade declarada de ampicilina se dissolvem em 45 minutos.
ENSAIOS DE PUREZA
Perda por dessecação. Proceder conforme descrito em Dessecação de substâncias antibióticas (V.5.2.17-4-Método 1). No mínimo 10,0% e, no máximo, 15,0%.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir, usando amostragem de, pelo menos, 10 cápsulas.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos
(V.5.2.17). Dissolver o padrão de ampicilina em água estéril, de modo a obter solução na concentração de, aproximadamente, 0,1 mg/ml. Remover o conteúdo das cápsulas e pesar exatamente.
Misturar os conteúdos. Transferir quantidade representativa da amostra, exatamente pesada, para balão volumétrico e diluir com tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) para obter solução de trabalho na concentração da solução padrão. Agitar de 3 a 5 minutos. Diluir soluções padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Dissolver o padrão de ampicilina em água, até concentração, aproximada, de 1,25 mg/ml. Remover o conteúdo das cápsulas e pesar exatamente. Misturar os conteúdos. Transferir quantidade representativa da amostra, exatamente pesada, para balão volumétrico e diluir com água de modo a obter concentração idêntica à do padrão. Agitar de 3 a 5 minutos. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar prova em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções, adicionadas de 10 ml de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
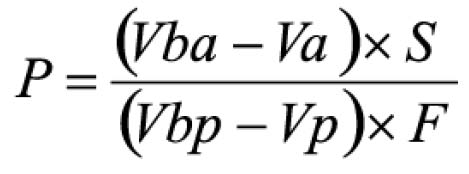
Em que
P = potência da amostra (mg/cápsula);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (mg/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes perfeitamente fechados, a temperatura inferior a 30ºC.
ROTULAGEM
Observar a legislação vigente.
_________________________________________________
XII.4 TAMPÕES
Tampão de sulfato de cobre
Preparação: no momento de uso, misturar 15 ml de solução B em 985 ml de solução A.
Solução A: dissolver 15,22 g de fosfato (dibásico) de sódio anidro em
quantidade suficiente de água. Adicionar 9,75 g de ácido cítrico
monoidratado e completar o volume para 1000 ml com água. Acertar
a pH 5,2 com auxílio de hidróxido de sódio ou ácido cítrico.
Solução B: dissolver 0,313 g de sulfato de cobre pentaidratado em
100 ml de água.
79.2
AMPICILINA TRIIDRATADA COMPRIMIDOS
Comprimidos de ampicilina triidratada são constituídos de ampicilina com um ou mais agentes diluentes, lubrificantes e conservantes adequados.
A potência é de, no mínimo, 90,0% e, no máximo, 120,0% do valor declarado de ampicilina.
IDENTIFICAÇÃO
Proceder conforme descrito no teste B de Identificação na monografia
Ampicilina triidratada.
CARACTERÍSTICAS
Determinação de peso (V.1.1). Cumpre o teste.
Dureza (V.1.3.1). Cumpre o teste.
Friabilidade (V.1.3.2). Cumpre o teste.
Teste de desintegração (V.1.4.1). No máximo 15 minutos.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
TESTE DE DISSOLUÇÃO (V.1.5)
Meio de dissolução: água, 900 ml
Aparelhagem: cesta, 100 rpm
Tempo: 45 minutos
Procedimento: após o teste, retirar alíquotas do meio de dissolução, filtrar e diluir, em tampão de sulfato de cobre, até concentração adequada. Prosseguir conforme descrito no Teste de dissolução em Ampicilina cápsulas.
Tolerância: não menos que 75% da quantidade declarada de ampicilina se dissolvem em 45 minutos.
ENSAIOS DE PUREZA
Água (V.2.20). No mínimo, 9,5% e, no máximo, 12%.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos a seguir descritos, usando amostragem de, pelo menos, 10 comprimidos.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Dissolver o padrão de ampicilina em água estéril, de modo a obter solução na concentração de, aproximadamente, 0,1 mg/ml. Pesar e pulverizar os comprimidos. Transferir quantidade representativa da amostra, exatamente pesada, para frasco volumétrico e diluir com tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2) para obter solução de trabalho na concentração da solução padrão. Agitar de 3 a 5 minutos. Diluir soluções padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Dissolver o padrão de ampicilina emágua, até concentração, aproximada, de 1,25 mg/ml. Pesar e pulverizar
os comprimidos. Transferir quantidade representativa da
amostra, exatamente pesada, para balão volumétrico e diluir com água
de modo a obter concentração idêntica a do padrão. Agitar de 3 a 5
minutos. Transferir 2 ml da solução padrão para erlenmeyer com
tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar
e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico
1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de
tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3
gotas de solução de amido SI e prosseguir com a titulação até o
desaparecimento da cor azul. Proceder da mesma maneira com a
solução amostra. Realizar prova em branco, da amostra e do padrão,
por meio da titulação de 2 ml de ambas as soluções, adicionadas de
10 ml de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao
ponto final, adicionar 3 gotas de solução de amido SI e prosseguir
com a titulação até o desaparecimento da cor azul. Titular com tiossulfato
de sódio 0,01 M SV.
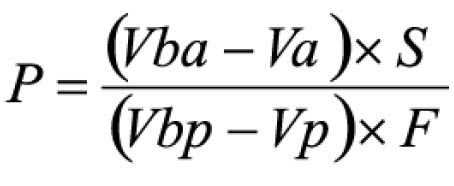
Em que
P = potência da amostra (mg/comprimido);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (mg/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes perfeitamente fechados, protegidos da umidade e a temperatura inferior a 30 ºC.
ROTULAGEM
Observar a legislação vigente.
_________________________________________________
XII.4 TAMPÕES
Tampão de sulfato de cobre
Preparação: no momento de uso, misturar 15 ml de solução B em 985 ml de solução A.
Solução A: dissolver 15,22 g de fosfato (dibásico) de sódio anidro em
quantidade suficiente de água. Adicionar 9,75 g de ácido cítrico
monoidratado e completar o volume para 1000 ml com água. Acertar
a pH 5,2 com auxílio de hidróxido de sódio ou ácido cítrico.
Solução B: dissolver 0,313 g de sulfato de cobre pentaidratado em
100 ml de água.
79.3
AMPICILINA TRIIDRATADA PÓ PARA SUSPENSÃO INJETÁVEL
Ampicilina triidratada estéril para suspensão é mistura seca de ampicilina triidratada com um ou mais agentes adequados para suspensão, tampões, estabilizantes e conservantes. A potência é de, no mínimo, 90,0% e, no máximo, 120,0% do valor declarado de ampicilina triidratada.
IDENTIFICAÇÃO
Proceder conforme descrito no teste B de Identificação na monografia
Ampicilina triidratada.
CARACTERÍSTICAS
pH (V.2.19). 5,0 a 7,0. Após reconstituição com o diluente.
Determinação do peso (V.1.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
ENSAIOS DE PUREZA
Perda por dessecação. Proceder conforme descrito em Dessecação de substâncias antibióticas (V.5.2.17-4-Método 2). No mínimo, 11,4% e, no máximo, 14,0%.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1-5). Cumpre o teste. Empregar o método de filtração por membrana. Dissolver 6 g da amostra em 800 ml de Fluido II contendo quantidade suficiente de penicilinase estéril para inativar a ampicilina, agitar até total solubilização e proceder como descrito.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 1,0 ml/kg, empregando solução de ampicilina, na concentração de 20 mg/ml, em água isenta de pirogênios.
Endotoxinas bacterianas (V.5.1.9). No máximo 0,15 UE/mg de ampicilina.
DETERMINAÇÃO DA POTÊNCIA
Para determinação da potência de pó para solução injetável de ampicilina, empregar um dos métodos descritos a seguir, utilizando amostragem mínima de 10 frascos.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Dissolver padrão e amostra em água estéril de modo a obter solução na concentração de 0,1 mg/ml. Diluir solução padrão e amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução 2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Dissolver e diluir padrão e amostra reconstituída em água, até concentração de, aproximadamente, 1,25 mg/ml. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder ao mesmo ensaio com a solução amostra. Realizar prova em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções, adicionadas de 10 ml da solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
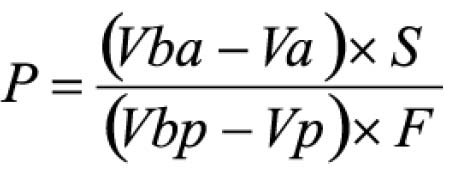
Em que
P = potência da amostra (mg/frasco-ampola);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (mg/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 30ºC.
ROTULAGEM
Observar a legislação vigente.
79.4
AMPICILINA TRIIDRATADA PÓ PARA SUSPENSÃO ORAL
Ampicilina pó para suspensão oral é mistura de ampicilina triidratada com um ou mais agentes corantes, aromatizantes, tampões, adoçantes e conservantes. A potência é de, no mínimo, 90,0% e, no máximo,120,0% do valor declarado de ampicilina.
IDENTIFICAÇÃO
Proceder conforme descrito no teste B de Identificação na monografia
Ampicilina triidratada.
CARACTERÍSTICAS
pH (V.2.19). 5,0 a 7,5. Determinar na suspensão reconstituída, conforme indicação do produtor.
Determinação do peso (V.1.1). Cumpre o teste.
Determinação do volume (V.1.2). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
ENSAIOS DE PUREZA
Água (V.2.20.1). No máximo 5,0%, em quantidade contendo o equivalente a 100 mg/ml de ampicilina após a reconstituição.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Dissolver o padrão de ampicilina em água estéril, de modo a obter solução na concentração de, aproximadamente, 0,1 mg/ml. Reconstituir o conteúdo conforme indicado pelo produtor.
Transferir quantidade representativa da amostra para balão volumétrico
e diluir com tampão fosfato de potássio 0,1 M, estéril, pH 8,0(Solução 2) para obter solução de trabalho na concentração da solução
padrão. Agitar de 3 a 5 minutos. Diluir solução padrão e
amostra, em tampão fosfato de potássio 0,1 M, estéril, pH 8,0 (Solução
2), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Dissolver o padrão de ampicilina emágua, até concentração, aproximada, de 1,25 mg/ml. Reconstituir o conteúdo conforme indicado pelo produtor. Transferir quantidade representativa da amostra para frasco volumétrico e diluir com água de modo a obter concentração idêntica à do padrão. Agitar de 3 a 5 minutos. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar provas em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções, adicionadas de 10 ml de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
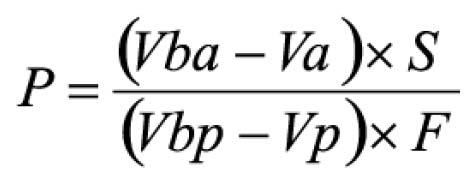
Em que
P = potência da amostra (mg/frasco);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (mg/ml);
F = fator de diluição da amostra.
TESTES DE SEGURANÇA BIOLÓGICA
Contagem de microrganismos viáveis (V.5.1.6.1). Bactérias aeróbicas totais: no máximo 1000 UFC/ml. Fungos e leveduras: no máximo 100 UFC/ml.
Pesquisa e identificação de patógenos (V.5.1.7). Cumpre o teste.
EMBALAGEM E ARMAZENAMENTO
Em recipientes bem-fechados, protegidos da umidade e a temperatura inferior a 30 ºC.
ROTULAGEM
Observar a legislação vigente.
82
BENZILPENICILINA BENZATINA
Benzylpenicillinum benzathinum
(C16H18N2O4S)2.C16H20N2
909,13 0 111.02- 3
Sal composto do ácido [2S-(2?,5?,6?)]-3,3-dimetil-7-oxo-6-[(fenilacetil) amino]-4-tia-1-azabiciclo[3.2.O]heptano-2-carboxílico com a N, N'-bis(fenilmetil)-1,2-etano-diamina
Apresenta potência de, no mínimo, 1 090 Unidades e, no máximo, 1
272 Unidades de benzilpenicilina por miligrama.
DESCRIÇÃO
Caracteres físicos. Pó cristalino branco.
Solubilidade. Muito pouco solúvel em água, facilmente solúvel em dimetilformamida e formamida, pouco solúvel em etanol, muito pouco solúvel em clorofórmio, praticamente insolúvel em éter.
IDENTIFICAÇÃO
O teste de identificação A pode ser omitido se forem realizados os testes B, C e D. Os testes de identificação B, C e D podem ser omitidos se for realizado o teste A.
A. O espectro de absorção no infravermelho (V.2.14-4) da amostra, dispersa em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de benzilpenicilina benzatina padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando como suporte sílica-gel H, e como fase móvel mistura de acetona-acetato de amônio a 15,4% (p/V) (30:70), pH 7,0ajustado com amônia. Aplicar, separadamente à placa, 1 ml de cada uma das seguintes soluções.
Solução (1): solução a 0,5% (p/V) da amostra em metanol.
Solução (2): solução a 0,5% (p/V) do padrão em metanol.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e expor a vapores de iodo até o aparecimento das manchas. Examinar sob luz visível. A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (2).
C. Transferir cerca de 2 mg da amostra para tubo de ensaio. Umedecer com 0,05 ml de água e adicionar 2 ml da mistura de 2 ml de solução de formaldeído com 100 ml de ácido sulfúrico. Agitar o tubo e observar a cor. Deve-se apresentar quase incolor. Imergir o tubo em banho-maria durante um minuto. Desenvolve-se coloração marromavermelhada.
D. Adicionar 2 ml de hidróxido de sódio a 0,1 g de amostra e agitar por 2 minutos. Agitar a mistura duas vezes, cada uma com 3 ml deéter etílico. Reunir as camadas etéreas e evaporar até a secura. Dissolver o resíduo em 1 ml de etanol a 50% (V/V). Adicionar 5 ml de solução de ácido pícrico a 1,0% (p/V), aquecer à temperatura de 90 0C, durante 5 minutos, e deixar esfriar lentamente. Separar os cristais e recristalizar com etanol a 25% (V/V) contendo 1% (p/V) de ácido pícrico. Os cristais fundem-se à temperatura de, aproximadamente, 214 0C.
ENSAIOS DE PUREZA
pH (V.2.19). 4,0 a 6,5. Determinar em solução obtida dissolvendo 0,05 g de amostra em 50 ml de etanol absoluto, adicionando 50 ml deágua.
Água (V.2.20.1). 5,0 a 8,0%. Determinar em 0,3 g de amostra.
Cristalinidade. Suspender algumas partículas da amostra em óleo mineral, transferir para uma lâmina de vidro e examinar por meio de microscópio dotado de luz polarizada. As partículas exibem birrefringência, que se extingue ao movimentar a amostra por meio de ajuste micrométrico.
TEOR DE BENZATINA
Teor de benzatina. 24,0 a 27,0%, calculado sobre a substância anidra.
Pesar cerca de 1 g de benzilpenicilina benzatina e adicionar 30 ml de solução saturada de cloreto de sódio e 10 ml de solução de hidróxido de sódio a 20% (p/V). Agitar e extrair, quatro vezes, com 50 ml deéter etílico. Lavar a fase etérea reunida, três vezes, com 10 ml deágua.
Reunir as águas de lavagem e extrair com 25 ml de éter etílico.
Juntar esta camada etérea a anteriormente reunida. Reduzir a solução de éter, por meio de evaporação, para volume aproximado de 5 ml.
Adicionar 2 ml de etanol absoluto e evaporar a secura. Dissolver o resíduo em 50 ml de ácido acético glacial. Titular com ácido perclórico 0,1 M, empregando 1 ml de solução de 1-naftolbenzeína a 1% em ácido acético glacial. Realizar titulação em branco. 1 ml de ácido perclórico 0,1 M é equivalente a 12,02 mg de C16H20N2.
TESTES DE SEGURANÇA BIOLÓGICA
Benzilpenicilina benzatina destinada a preparação parenteral deve cumprir com os seguintes testes adicionais:
Esterilidade (V.5.1.1-4). Cumpre o teste. Empregar o método de inoculação ou direto, usando meio de tioglicolato fluido e meio caseínasoja contendo polissorbato 80. Adicionar penicilinase estéril em quantidade suficiente para neutralizar a benzilpenicilina em cada tubo.
Agitar os frascos diariamente.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 0,5 ml/kg, empregando solução de benzilpenicilina benzatina, na concentração de 4 000 U/ml, em solução salina livre de pirogênios.
Endotoxinas bacterianas (V.5.1.9). No máximo 0,01 UE/100 unidades de benzilpenicilina.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos abaixo.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Adicionar metanol ao padrão e amostra até completa dissolução. Diluir, com solução tampão fosfato de potássio 1%, estéril, pH 6,0 (solução 1), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Diluir padrão de benzilpenicilina potássica e amostra até concentração de, aproximadamente, 2 000 U/ml, utilizando solução tampão fosfato de potássio a 1%, pH 6,0 (solução 1), não-estéril. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M.
Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI, e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar provas em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas soluções, adicionadas de 10 ml da solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M.
Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
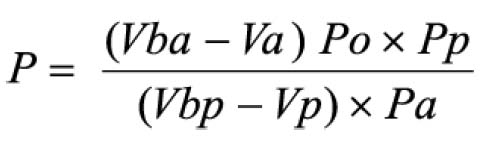
Em que
P = potência da amostra (U/mg);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
Pó = potência do padrão (U/mg);
Pp = peso do padrão (mg);
Pa = peso da amostra (mg).
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, à temperatura inferior a 30 oC. Sendo destinado a produção de formas farmacêuticas injetáveis, deverá ser embalado em recipientes estéreis.
CLASSE TERAPÊUTICA
Antibiótico.
82.1
BENZILPENICILINA BENZATINA ESTÉRIL PÓ PARA SUSPENSÃO INJETÁVEL
Benzilpenicilina benzatina estéril para suspensão injetável é mistura seca de benzilpenicilina benzatina com um ou mais agentes adequados para suspensão ou dispersão e, contendo ou não, um ou mais conservantes e substâncias tampão. A potência é de, no mínimo, 90,0% e, no máximo, 115,0% do valor declarado de benzilpenicilina.
IDENTIFICAÇÃO
Proceder conforme descrito nos testes de identificação na monografia
Benzilpenicilina benzatina.
CARACTERÍSTICAS
pH (V.2.19). 5,0 a 7,5. Após reconstituição com o diluente.
Determinação de peso (V.1.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
ENSAIOS DE PUREZA
Água (V.2.20.1). 5,0 a 8,0%. Determinar em 0,3 g de amostra.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1-4). Cumpre o teste. Empregar o método de inoculação ou direto, usando meio de tioglicolato fluido e meio caseínasoja contendo polissorbato 80. Adicionar penicilinase estéril em quantidade suficiente para neutralizar a benzilpenicilina em cada tubo.
Agitar os frascos diariamente.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 0,5 ml/kg, empregando solução de benzilpenicilina benzatina, na concentração de 4 000 U/ml em solução salina livre de pirogênios.
Endotoxinas bacterianas (V.5.1.9). No máximo 0,01 UE/100 unidades de benzilpenicilina.
DETERMINAÇÃO DA POTÊNCIA
Para determinação da potência da benzilpenicilina benzatina, empregar um dos métodos descritos a seguir, utilizando amostragem mínima de 10 frascos.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Adicionar metanol à suspensão até completa dissolução. Diluir, amostra e padrão, com solução tampão fosfato de potássio a 1%, estéril, pH 6,0 (tampão 1), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Diluir padrão de benzilpenicilina potássica e amostra reconstituída, até concentração de, aproximadamente, 2000 U/ml de benzilpenicilina benzatina, utilizando solução tampão fosfato de potássio a 1%, pH 6,0 (tampão 1) não estéril.Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar provas em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas soluções, adicionadas de 10 ml de solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
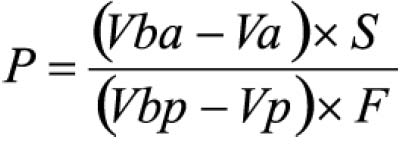
Em que
P = potência da amostra (U/frasco-ampola);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (U/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 30 oC.
ROTULAGEM
Observar a legislação vigente.
83
BENZILPENICILINA POTÁSSICA
Benzylpenicillinum kalicum
Sal monopotássico do ácido [2S-(2α,5α,6β)]-3,3-dimetil-7-oxo-6-
[(fenilacetil)amino]-4-tia-1-azabiciclo[3.2.0]heptano-2-carboxílico.
Apresenta potência de, no mínimo, 1 440 Unidades e de, no máximo,
1 680 Unidades de benzilpenicilina por miligrama.
DESCRIÇÃO
Caracteres Físicos. Pó cristalino branco ou quase branco.
Solubilidade. Muito solúvel em água, praticamente insolúvel em clorofórmio,éter, óleos e parafina líquida.
Constantes físico-químicas
Poder rotatório específico (V.2.8): + 270º a + 300º, determinado em solução a 2% (p/V) em água isenta de dióxido de carbono, calculado em relação à substância dessecada.
IDENTIFICAÇÃO
O teste de identificação A pode ser omitido se forem realizados os testes B, C e D. Os testes de identificação B e C podem ser omitidos se forem realizados os testes A e D.
A. O espectro de absorção no infravermelho (V.2.14-4) de amostra dispersa em brometo de potássio apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de benzilpenicilina potássica padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando como suporte sílica-gel H e como fase móvel mistura de acetona-acetato de amônio a 15,4% (p/V) (30:70), pH 5,0, ajustado com ácido acético glacial. Aplicar, separadamente, à placa 1,0 ml de cada uma das seguintes soluções.
Solução (1): solução a 0,5% (p/V) da amostra em água.
Solução (2): solução a 0,5% (p/V) do padrão em água.
Solução (3): solução contendo 0,5% (p/V) do padrão e 0,5% (p/V) de fenoximetilpenicilina potássica padrão em água.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e expor a vapores de iodo até o aparecimento das manchas. Examinar sob luz visível. A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução(2). As manchas obtidas com as soluções (2) e (3) não devem ser correspondentes.
C. Transferir cerca de 2 mg da amostra para tubo de ensaio. Umedecer com 0,05 ml de água e adicionar 2 ml da mistura de 2 ml de solução de formaldeído com 100 ml de ácido sulfúrico. Agitar o tubo e observar a cor. Deve-se apresentar quase incolor. Imergir o tubo em banho-maria fervente durante um minuto. Desenvolve-se coloração marrom-avermelhada.
D. Responde à reação de identificação para íons potássio. (V.3.1.1- 5).
ENSAIOS DE PUREZA
pH (V.2.19). 5,5 a 7,5. Determinar em solução aquosa, isenta de dióxido de carbono, a 6% (p/V).
Perda por dessecação. Proceder conforme descrito em Dessecação de substâncias antibióticas (V.5.2.17-4-Método 2). No máximo 1,0%.
Cristalinidade. Suspender algumas partículas da amostra em óleo mineral, transferir para uma lâmina de vidro e examinar por meio de microscópio dotado de luz polarizada. As partículas exibem birrefringência, que se extingue ao movimentar a amostra por meio de ajuste micrométrico.
TESTES DE SEGURANÇA BIOLÓGICA
Benzilpenicilina potássica destinada à preparação parenteral deve cumprir com os seguintes testes adicionais:
Esterilidade (V.5.1.1-5). Cumpre o teste. Empregar o método de filtração por membranas.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 1,0 ml/kg, empregando solução de benzilpenicilina, na concentração de 20 000 U/ml em solução salina livre de pirogênio.
Endotoxinas bacterianas (V.5.1.9). No máximo 0,01 UE/100 unidades de benzilpenicilina.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Diluir com solução tampão fosfato de potássio a 1%, estéril, pH 6,0 (solução 1), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Diluir amostra e padrão até concentração de, aproximadamente, 2 000 U/ml de benzilpenicilina potássica utilizando solução tampão fosfato de potássio a 1%, pH 6,0 (solução 1), não estéril. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso por 15 minutos ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar provas em branco da amostra e do padrão, através da titulação de 2 ml da ambas as soluções, adicionadas de 10 ml da solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M.
Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
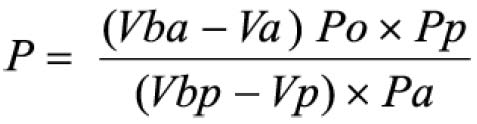
P = potência da amostra (U/mg);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
Pó = potência do padrão (U/mg);
Pp = peso do padrão (mg);
Pa = peso da amostra (mg).
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 30 oC. Sendo destinado à produção de formas farmacêuticas injetáveis, deverá ser embalado em recipientes estéreis.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antibiótico.
83.1
BENZILPENICILINA POTÁSSICA ESTÉRIL PÓ PARA SOLUÇÃO INJETÁVEL
Benzilpenicilina potássica estéril para preparação parenteral é mistura seca de benzilpenicilina potássica e citrato de sódio como tampão, em quantidade não inferior a 4% e não-superior a 5% da fase sólida total.
Pode ser empregado ácido cítrico, em quantidade não-superior a 0,15% da fase sólida total em substituição ao citrato de sódio. A potência é de, no mínimo, 90,0% e, no máximo, 120,0% do valor declarado de benzilpenicilina.
IDENTIFICAÇÃO
Proceder conforme descrito nos testes de identificação da monografia Benzilpenicilina potássica.
CARACTERÍSTICAS
pH (V.2.19). 6,0 a 8,5. Determinar após reconstituição da amostra com o diluente.
Determinação de peso (V.1.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
ENSAIOS DE PUREZA
Perda por dessecação. Proceder conforme descrito em Dessecação de substâncias antibióticas (V.5.2.17-4-Método 2). No máximo 1,5%.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1-5). Cumpre o teste. Empregar o método de filtração por membranas.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 1,0 ml/kg, empregando solução de benzilpenicilina, na concentração de 20 000 U/ml, em solução salina, livre de pirogênio.
Endotoxinas bacterianas (V.5.1.9). No máximo 0,01 UE/100 unidades de benzilpenicilina.
DETERMINAÇÃO DA POTÊNCIA
Para determinação da potência da benzilpencilina potássica, empregar um dos métodos descritos a seguir, utilizando amostragem mínima de 10 frascos.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Diluir, amostra e padrão, com solução tampão fosfato de potássio a 1%, estéril, pH 6,0 (solução 1), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Diluir amostra reconstituída e padrão, até concentração de, aproximadamente, 2 000 U/ml de benzilpenicilina potássica, utilizando solução tampão de fosfato de potássio a 1%, pH 6,0 (solução 1) não estéril. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos.
Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar prova em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas soluções adicionadas de 10 ml de solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
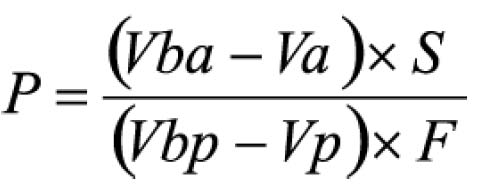
Em que
P = potência da amostra (U/frasco-ampola);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (U/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 30 oC.
ROTULAGEM
Observar a legislação vigente.
84
BENZILPENICILINA PROCAÍNA
Benzylpenicillinum procainum
C16H18N2O4S.C13H20N202. H2O588,72 0 111.04- X
Sal monoidratado composto do ácido [2S-(2α,5α,6β)]-3,3-dimetil-7- oxo-6-[(fenilacetil)amino]-4-tia-1-azabiciclo [3.2.0]heptano-2-carboxílico com o 2-(dietilamino)etil 4-aminobenzoato (1:1)
Apresenta potência de, no mínimo, 900 Unidades e, no máximo, 1 050 Unidades de benzilpenicilina por miligrama, e não-menor que 3D9E,8S%CR eI ÇnÃãoO mais que 42,0% de C13H20N2O2.
Caracteres Físicos. Pó cristalino branco.
Solubilidade. Pouco solúvel em água, ligeiramente solúvel em etanol e clorofórmio.
Constantes físico-químicas
Poder rotatório específico (V.2.8): + 165o a + 180o. Determinado em solução a 1,0% (p/V) da mistura de água e acetona (2:3).
IDENTIFICAÇÃO
O teste de identificação A pode ser omitido se forem realizados os testes B, C e D. Os testes de identificação B, C e D podem ser omitidos se for realizado o teste A.
A. O espectro de absorção no infravermelho (V.2.14-4) da amostra, dispersa em brometo de potássio, apresenta máximos de absorção nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de benzilpenicilina procaína padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando sílica-gel H, como suporte, e como fase móvel mistura de acetona-acetato de amônio a 15,4% (p/V) (30:70), pH 7,0 ajustado com amônia. Aplicar separadamente à placa, 1,0 ?l de cada uma das seguintes soluções:
Solução (1): solução 0,5% (p/V) da amostra em acetona.
Solução (2): solução 0,5% (p/V) do padrão em acetona.
Desenvolver o cromatograma. Remover a placa, deixar secar ao ar e expor a vapores de iodo até o aparecimento das manchas. Examinar sob luz visível. A mancha principal obtida com a solução (1) corresponde em posição, cor e intensidade àquela obtida com a solução (2).
C. Transferir cerca de 2 mg da amostra para tubo de ensaio. Umedecer com 0,05 ml de água e adicionar 2 ml da mistura de 2 ml de solução de formaldeído com 100 ml de ácido sulfúrico. Agitar o tubo e observar a cor. Deve-se apresentar quase incolor. Imergir o tubo em banho-maria durante um minuto. Desenvolve-se coloração marromavermelhada.
D. Adicionar 2 ml de ácido clorídrico 2 M a 0,1 g de amostra. A solução deverá fornecer reações de identificação de Amina aromática primária (V.3.1.1).
ENSAIOS DE PUREZA
pH (V.2.19). 5,0 a 7,5. Determinar em solução obtida dissolvendo 0,05 g da amostra em 15 ml de água e agitando até completa dissolução.
Água (V.2.20.1). 2,8 a 4,2%. Determinar em 0,5 g da amostra.
Cristalinidade. Suspender algumas partículas da amostra em óleo mineral, transferir para uma lâmina de vidro e examinar por meio de microscópio dotado de luz polarizada. As partículas exibem birrefringência, que se extingue ao movimentar a amostra por meio de ajuste micrométrico.
TEOR DE PROCAÍNA
Por espectrofotometria de absorção no visível (V.2.14-3). Dissolver a amostra em tampão fosfato de potássio 1%, pH 6,0 (solução 1) não estéril, e diluir, no mesmo solvente, até obter concentração de 60 U/ml. Dissolver 27,55 mg de cloridrato de procaína padrão em 1 000 ml do tampão fosfato pH 6,0 (cada ml desta solução equivale a 60 unidades de benzilpenicilina procaína). Transferir 1,0 ml das soluções amostra e padrão para balões volumétricos de 25 ml. Adicionar, a cada balão, 0,5 ml de ácido clorídrico 4 M e 1,0 ml de nitrito de sódio 0,1% (p/V). Agitar e deixar em contato por 2 minutos. Adicionar 1,0 ml de sulfamato de amônio 0,5% (p/V), agitar e deixar em contato por 2 minutos. Adicionar 1,0 ml de dicloridrato de N-1- naftiletilendiamina 0,1% (p/V), agitar e deixar em contato por 2 minutos. Completar os volumes com água destilada. Realizar preparação do branco, em paralelo, nas mesmas condições, omitindo-se a adição de soluções amostra e padrão. Medir as absorvâncias das soluções em 540 nm, utilizando a preparação do branco para zerar o aparelho. Calcular o teor de C10H10N4O2S na amostra com base nas lTeEitSuTraEs So bDtiEd aSsE. GURANÇA BIOLÓGICA Benzilpenicilina procaína destinada à preparação parenteral deve cumprir com os seguintes testes adicionais:
Esterilidade (V.5.1.1-5). Cumpre o teste. Empregar o método de filtração por membranas.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 2 ml/kg, empregando solução de benzilpenicilina, na concentração de 2 000 U/ml em solução salina livre de pirogênio.
Endotoxinas bacterianas (V.5.1.9). No máximo 0,01 UE/100 unidades de benzilpenicilina.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Preparar amostra e padrão através de diluição em solução tampão fosfato de potássio a 1%, estéril, pH 6,0 (solução 1).
B. Por método iodométrico. Diluir padrão de benzilpenicilina e amostra até concentração de, aproximadamente, 2 000 U/ml, utilizando solução tampão fosfato de potássio a 1%, pH 6,0 (solução 1), nãoestéril.
Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar prova em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas soluções, adicionadas de 10 ml da solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
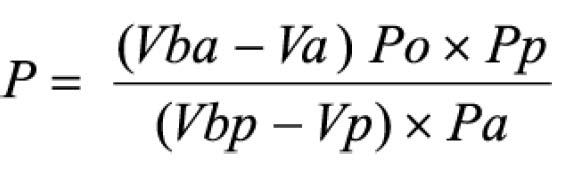
Em que
P = potência da amostra (U/mg);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
Pó = potência do padrão (U/mg);
Pp = peso do padrão (mg);
Pa = peso da amostra (mg).
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 30 oC. Sendo destinado à produção de formas farmacêuticas injetáveis, deverá ser embalada em recipientes estéreis.
ROTULAGEM
Observar a legislação vigente.
CLASSE TERAPÊUTICA
Antibiótico.
84.1
BENZILPENICILINA PROCAÍNA ESTÉRIL PÓ PARA SUSPENSÃO INJETÁVEL
Benzilpenicilina procaína estéril para suspensão injetável é mistura seca de benzilpenicilina procaína com um ou mais agentes adequados para suspensão ou dispersão, contendo ou não conservantes e substâncias tampão. A potência é de, no mínimo, 90,0% e, no máximo, 115,0% do valor declarado de benzilpenicilina.
IDENTIFICAÇÃO
Proceder conforme descrito nos testes de identificação da monografia
Benzilpenicilina procaína.
CARACTERÍSTICAS
pH (V.2.19). 5,0 a 7,5. Determinar após reconstituição com diluente.
Determinação de peso (V.1.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
ENSAIOS DE PUREZA
Água (V.2.20.1). 2,8 a 4,2%. Determinar em 0,5 g da amostra.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1-5). Cumpre o teste. Empregar o método de filtração por membrana.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 2,0 ml/kg, empregando solução de benzilpenicilina, na concentração de 2 000 U/ml, em solução salina livre de pirogênio.
Endotoxinas bacterianas (V.5.1.9). No máximo 0,01 UE/100 unidades de benzilpenicilina.
DETERMINAÇÃO DA POTÊNCIA
Para determinação da potência da benzilpencilina procaína, empregar um dos métodos descritos a seguir, utilizando amostragem mínima de 10 frascos.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Diluir, amostra e padrão, com solução tampão fosfato de potássio a 1%, estéril, pH 6,0 (solução 1), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Diluir padrão de benzilpenicilina potássica e amostra reconstituída, até concentração de, aproximadamente, 2 000 U/ml de benzilpenicilina procaína, utilizando solução tampão de fosfato de potássio a 1%, pH 6,0 (solução 1) não-estéril.
Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar provas em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas soluções, adicionadas de 10 ml de solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
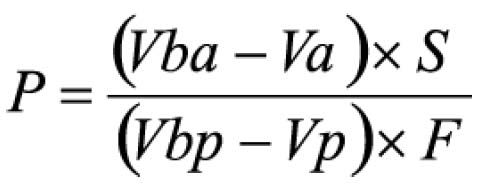
Em que
P = potência da amostra (U/frasco-ampola);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (U/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 30 oC.
ROTULAGEM
Observar a legislação vigente.
85
BENZILPENICILINA SÓDICA
Benzylpenicillinum natricum
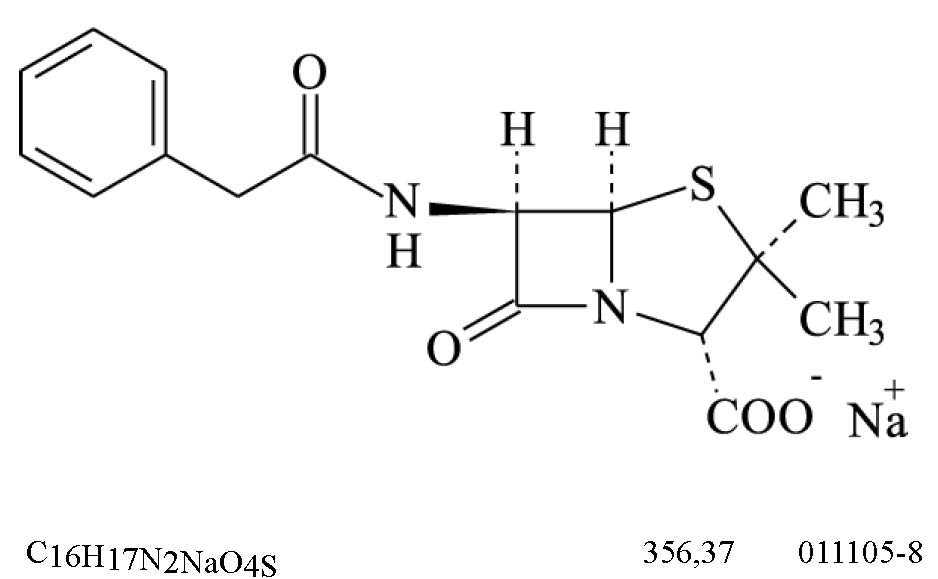
Sal monossódico do ácido [2S-(2α,5α,6β)-3,3-dimetil-7-oxo-6-[(fenilacetil amino)]-4-tia-1-azabiciclo[3.2.0]heptano-2-carboxílico
Apresenta potência de, no mínimo, 1 500 Unidades e, no máximo, 1 por miligrama.
Caracteres físicos. Pó cristalino branco ou quase branco.
Solubilidade. Muito solúvel em água, éter, óleos e parafina líquida.
Constantes físico-químicas
Poder rotatório específico (V.2.8): + 285º a +310º. Determinado em solução a 0,5% (p/V) em água livre de dióxido de carbono, calculado em relação à substância dessecada.
IDENTIFICAÇÃO
O teste de identificação A pode ser omitido se forem realizados os testes B, C e D. Os testes de identificação B e C podem ser omitidos se forem realizados os testes A e D.
A. O espectro de absorção no infravermelho (V.2.14-4) da amostra, dispersa em brometo de potássio, apresenta máximos de absorção somente nos mesmos comprimentos de onda e com as mesmas intensidades relativas daqueles observados no espectro de benzilpenicilina sódica padrão, preparado de maneira idêntica.
B. Proceder conforme descrito em Cromatografia em camada delgada (V.2.17.1), utilizando como suporte sílica-gel H e como fase móvel mistura de acetona-acetato de amônio a 15,4% (p/V) (30:70), pH 5,0 ajustado com ácido acético glacial. Aplicar, separadamente, à placa, 1 de cada uma das seguintes soluções.
Solução (1): solução 0,5% (p/V) da amostra em metanol.
Solução (2): solução 0,5% (p/V) do padrão em metanol.
Desenvolver o cromatograma. Remover a placa e deixar secar ao ar e expor a vapores de iodo até o aparecimento das manchas. Examinar sob luz visível. A mancha principal obtida com a solução (1) deve ser semelhante em posição, cor e intensidade àquela obtida com a solução (2).
C. Transferir cerca de 2 mg da amostra para tubo de ensaio. Umedecer com 0,05 ml de água e adicionar 2 ml da mistura de 2 ml de solução de formaldeído com 100 ml de ácido sulfúrico. Agitar o tubo e observar a cor. Deve-se apresentar quase incolor. Imergir o tubo em banho-maria durante um minuto. Desenvolve-se coloração marromavermelhada.
D. A substância responde à reação de identificação para íons sódio ( V. 3.1).
ENSAIOS DE PUREZA
pH (V.2.19). 5,5 a 7,5. Determinar em solução a 10% (p/V) emágua.
Perda por dessecação. Proceder conforme descrito em Dessecação de substâncias antibióticas (V.5.2.17-4-Método 2). No máximo 1,5%.
Cristalinidade. Suspender uma pequena porção da amostra, em óleo mineral e examinar por meio de microscópio dotado de luz polarizada.
As partículas exibem birrefringência, que se extingue ao movimentar a amostra por meio de ajuste micrométrico.
TESTES DE SEGURANÇA BIOLÓGICA
Benzilpenicilina sódica destinada à preparação parenteral deve cumprir os seguintes testes adicionais:
Esterilidade (V.5.1.1-5). Cumpre o teste. Empregar o método de filtração por membrana.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 1,0 ml/kg, empregando solução de benzilpenicilina, na concentração de 20 000 U/ml, em solução salina livre de pirogênio.
Endotoxinas bacterianas (V.5.1.9). No máximo 0,01 UE/100 unidades de benzilpenicilina.
DETERMINAÇÃO DA POTÊNCIA
Empregar um dos métodos descritos a seguir.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Preparar amostra e padrão através de diluição em solução tampão fosfato de potássio a 1%, estéril, pH 6,0 (solução 1).
B. Por método iodométrico. Diluir padrão de benzilpenicilina potássica e amostra até concentração de, aproximadamente, 2 000 U/ml, utilizando solução tampão fosfato de potássio a 1%, pH 6,0 (solução 1), não-estéril. Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M.
Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso por 15 minutos ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder ao mesmo ensaio com a solução amostra. Realizar a determinação dos brancos, da amostra e do padrão, por meio da titulação de 2 ml de ambas as soluções adicionadas de 10 ml da solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
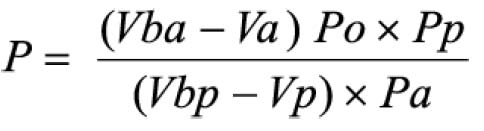
Em que
P = potência da amostra (U/mg);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
Pó = potência do padrão (U/mg);
Pp = peso do padrão (mg);
Pa = peso da amostra (mg).
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 30 oC. Sendo destinado à produção de formas farmacêuticas injetáveis, deverá ser embalado em recipientes estéreis.
ROTULAGEM
Observar a legislação vigente.
85.1
BENZILPENICILINA SÓDICA ESTÉRIL PÓ PARA SOLUÇÃO INJETÁVEL
Benzilpenicilina sódica estéril para preparação parenteral é mistura seca de benzilpenicilina sódica e citrato de sódio, como tampão, em quantidade não inferior a 4% e não superior a 5% da fase sólida total.
Pode ser empregado ácido cítrico, em quantidade não superior a 0,15% da fase sólida total em substituição ao citrato de sódio. A potência é de, no mínimo, 90,0% e, no máximo, 120,0% do valor declarado de benzilpenicilina.
IDENTIFICAÇÃO
Proceder conforme descrito nos testes de identificação da monografia Benzilpenicilina sódica.
CARACTERÍSTICAS
pH (V.2.19). 6,0 a 7,5. Determinar após reconstituição com diluente.
Determinação de peso (V.1.1). Cumpre o teste.
Uniformidade de doses unitárias (V.1.6). Cumpre o teste.
ENSAIOS DE PUREZA
Perda por dessecação. Proceder conforme descrito em Dessecação de substâncias antibióticas (V.5.2.17-4-Método 2). No máximo 1,0%.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade (V.5.1.1-5). Cumpre o teste. Empregar o método de filtração por membrana.
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 1,0 ml/kg, empregando solução de benzilpenicilina, na concentração de 20 000 U/ml, em solução salina livre de pirogênio.
Endotoxinas bacterianas (V.5.1.9). No máximo 0,01 UE/100 unidades de benzilpenicilina.
DETERMINAÇÃO DA POTÊNCIA
Para determinação da potência da benzilpencilina sódica, empregar um dos métodos descritos a seguir, utilizando amostragem mínima de 10 frascos.
A. Proceder conforme descrito em Ensaio microbiológico de antibióticos (V.5.2.17). Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Diluir, amostra e padrão, com solução tampão fosfato de potássio a 1%, estéril, pH 6,0 (solução 1), às concentrações empregadas na curva padrão.
B. Por método iodométrico. Reconstituir o conteúdo de 10 frascos conforme indicado pelo produtor. Diluir padrão de benzilpenicilina potássica e amostra reconstituída até concentração de, aproximadamente, 2 000 U/ml de benzilpenicilina sódica, utilizando solução tampão de fosfato de potássio a 1%, pH 6,0 (solução 1) não-estéril.
Transferir 2 ml da solução padrão para erlenmeyer com tampa esmerilhada e adicionar 2 ml de hidróxido de sódio M. Agitar e deixar em repouso por 15 minutos. Adicionar 2 ml de ácido clorídrico 1,2 M e 10 ml de solução de iodo 0,01 M SV. Deixar em repouso, por 15 minutos, ao abrigo da luz. Titular com solução de tiossulfato de sódio 0,01 M SV. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Proceder da mesma maneira com a solução amostra. Realizar prova em branco, da amostra e do padrão, por meio da titulação de 2 ml de ambas soluções, adicionadas de 10 ml de solução de iodo 0,01 M SV e 0,1 ml de ácido clorídrico M. Próximo ao ponto final, adicionar 3 gotas de solução de amido SI e prosseguir com a titulação até o desaparecimento da cor azul. Titular com tiossulfato de sódio 0,01 M SV.
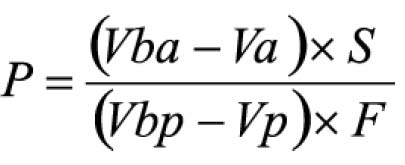
Em que
P = potência da amostra (U/frasco-ampola);
Vba = volume de titulante gasto na titulação do branco da amostra (ml);
Va = volume de titulante gasto na titulação da amostra (ml);
Vbp = volume de titulante gasto na titulação do branco do padrão (ml);
Vp = volume de titulante gasto na titulação do padrão (ml);
S = concentração do padrão (U/ml);
F = fator de diluição da amostra.
EMBALAGEM E ARMAZENAMENTO
Em recipientes hermeticamente fechados, a temperatura inferior a 30 oC.
ROTULAGEM
Observar a legislação vigente.
100
SOROS HIPERIMUNES PARA USO HUMANO
Immunosera ad usum humanum
Os soros hiperimunes são preparações contendo imunoglobulinas purificadas, de origem animal, que neutralizam especificamente toxinas bacterianas, bactérias, vírus ou componentes tóxicos do veneno de uma ou mais espécies de animais peçonhentos. Um conservante adequado pode ser adicionado e o produto final é apresentado sob forma líquida ou liofilizada. O produto líquido é límpido, incolor ou ligeiramente amarelado, não apresentando grumos ou partículas. O soro liofilizado consiste de pó branco ou ligeiramente amarelado que, uma vez reconstituído, apresenta as mesmas características do produto líquido.
As imunoglobulinas purificadas são obtidas por tratamento enzimático e precipitação fracionada, ou por outros procedimentos químicos ou físicos, de plasmas de animais sadios imunizados com os antígenos específicos. Durante o processo de imunização, os animais não devem ser tratados com penicilina ou estreptomicina.
Para assegurar a qualidade do produto nas diversas fases de processamento, devem ser realizados testes de esterilidade, pH, proteínas, atividade ou potência por métodos in vitro ou in vivo.
Antes do envase, amostras do produto são submetidas às determinações que seguem.
Cloreto de sódio. 0,70% a 0,90% (p/V).
Fenol. No máximo 0,35% (p/V).
Nitrogênio e proteínas. No máximo 0,3% (p/V) de nitrogênio nãoprotéico.
No máximo 15% (p/V) de proteínas.
Potência. É determinada de acordo com os procedimentos indicados nas monografias respectivas.
Sólidos totais. No máximo 20%.
Sulfato de amônio. No máximo 0,2% (p/V).
A preparação é distribuída assepticamente em ampolas ou frascosampola.
A liofilização do produto, quando requerida, deve assegurar concentração de água não superior a 1% do produto final.
IDENTIFICAÇÃO
A. Baseada na reação in vitro de antígeno-anticorpo por imunodifusão
duplo radial (Ouchterlony). Preparar gel de ágar a 1% (p/V) e distribuir
em lâmina para microscópio, de modo que resulte em fina
camada. Colocar em estufa a 37 ºC, sem secar. Adicionar volume de
4 ml de ágar na lâmina e colocar à temperatura de 2 ºC a 8 ºC em
câmara úmida por uma hora. Fazer orifícios no gel, mantendo a
mesma distância entre o orifício central e os periféricos. Preencher o
orifício central com solução do antígeno específico e os periféricos
com a amostra a testar, em diluições variáveis. Preencher um dos
orifícios com soro normal eqüino para controle negativo. Incubar a 37
ºC por 24 horas em câmara úmida e realizar a leitura em lâmpada
para contraste. Observar a presença de linha de precipitação, reação
de identidade entre os componentes analisados.
B. A Determinação da potência em animais suscetíveis pode ser utilizada para identificação do produto, conforme descrito na monografia da amostra correspondente.
CARACTERÍSTICAS
pH (V.2.19). 6,0 a 7,0.
Determinação de volume (V.1.2). Cumpre o teste.
ENSAIOS FÍSICO-QUÍMICOS
Cloreto de sódio. Em erlenmeyer de 50 ml, adicionar 10 ml da amostra diluída a 10% (V/V) em água bidestilada. Adicionar, com agitação, três gotas de solução difenilcarbazona-azul de bromofenol e, posteriormente, algumas gotas de ácido nítrico 0,2 M SV, até que a solução fique amarelo-esverdeada. Efetuar ensaio em branco. Titular com nitrato de mercúrio II 0,01 M SV, até o ponto de viragem, em que uma coloração violeta indica o ponto final. Cada ml de nitrato de mercúrio II 0,01 M SV equivale a 1,17 mg de NaCl. É facultado ao produtor a utilização do resultado obtido no produto antes do envase.
Entre 0,70 e 0,90% (p/V).
Fenol. Diluir a amostra de modo que a concentração de fenol esteja entre 5 ppm e 30 ppm. Adicionar 5 ml de tampão borato pH 9,0, 5 ml da solução de 4-aminofenazona a 0,1% (p/V) e 5 ml de solução aquosa de ferricianeto de potássio a 5% (p/V). Em paralelo, preparar branco e uma curva de calibração de fenol com concentrações variando de 5 ppm a 30 ppm. Proceder às leituras das absorvâncias da amostra, dos padrões e do branco a um comprimento de onda de 546 nm, 10 minutos após o término da reação. Utilizar a leitura dos padrões para fazer a curva de calibração e determinar a concentração de fenol na amostra por interpolação gráfica ou regressão linear. É facultado ao produtor a utilização do resultado obtido no produto antes do envase. No máximo 0,35% (p/V).
Nitrogênio protéico e proteínas (V.3.4.2). No máximo 0,3% (p/V) de nitrogênio não protéico e 15% (p/V) de proteínas. Para determinar a concentração de proteínas, multiplicar o resultado de nitrogênio protéico por 6,25. É facultado ao produtor a utilização do resultado obtido no produto antes do envase.
Sólidos totais. Em pesa-filtro previamente tarado, pesar exatamente 1 g da amostra em duplicata e colocar na capela de exaustão sobre placa de aquecimento, até a evaporação do líquido. Transferir o pesafiltro com a amostra para estufa a 105 ºC e deixar por 1 hora.
Transferir a amostra dessecada para dessecador, deixar por 30 minutos e pesar. Repetir o procedimento de dessecação até peso constante. Calcular a porcentagem de sólidos totais pela equação:
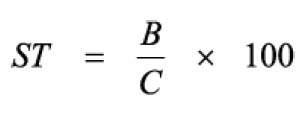
Em que
ST = % de sólidos totais;
B = diferença entre o pesa-filtro dessecado e o pesa-filtro vazio;
C = peso da amostra.
É facultado ao produtor a utilização do resultado obtido no produto antes do envase. No máximo 20%.
Sulfato de amônio. Diluir a amostra a 1% (V/V) com água bidestilada e transferir 10 ml da solução para tubo de Nessler. Transferir 1 ml de solução estoque de sulfato de amônio 0,6% (p/V) para balão volumétrico de 100 ml e completar o volume com água bidestilada.Diluir a solução em proporções 1:2, 1:3, 1:4 e transferir 10 ml de cada diluição para três tubos de Nessler. Adicionar 1 ml de reagente de Nessler a cada um dos tubos contendo a amostra e os padrões e comparar a cor. A intensidade da cor da amostra é igual ou menor que a da solução padrão diluída 1:3. É facultado ao produtor a utilização do resultado obtido no produto antes do envase. No máximo 0,2% (p/V).
| ensaio1º (0,4vn) | Repetição (0,4vn)2º | Repetição (0,8vn) | Resultado |
| - | |||
| + | satisfatório | ||
| + | - | satisfatório | |
| + | + | insatisfatório* | |
| + | + | - | satisfatório** |
| + | + | insatisfatório*** |
∗mesmo microrganismo isolado no ensaio e 1a repetição
∗∗ diferentes microrganismos isolados no ensaio e 1a repetição
∗∗∗presença de qualquer microrganismo
Pirogênios (V.5.1.2). Cumpre o teste. Injetar 1 ml/kg e não reutilizar os animais usados no teste.
DETERMINAÇÃO DA POTÊNCIA
Proceder conforme descrito na monografia específica. É facultado ao produtor a utilização do resultado obtido no produto antes do envase.
EMBALAGEM E ARMAZENAMENTO
A temperatura e o prazo de validade são os indicados pelo fabricante do soro, tendo como base evidências experimentais, aprovadas pela autoridade do controle nacional.
ROTULAGEM
Observar a legislação vigente.
_________________________________________________
XII.1 INDICADORES
Solução de difenilcarbazona-azul de bromofenol SI
Preparação - Em balão volumétrico de 25 ml, dissolver 12 mg de
difenilcarbazona e 12,5 mg de azul de bromofenol em 15 ml d
etanol. Completar o volume com etanol e acondicionar a solução em
frasco âmbar à temperatura de 4 ºC a 8 ºC.
XII.4 TAMPÕES
Tampão borato - pH 9,0
Preparação - Misturar 1 000 ml da solução A com 420 ml da solução B, preparadas como se segue.
Solução A: dissolver 6,18 g de ácido bórico em solução de cloreto de potássio 0,1 M SV e completar volume para 1 000 ml com a mesma solução.
Solução B: dissolver 2 g de hidróxido de sódio em água e completar o volume para 500 ml com o mesmo solvente.
117
VACINA BCG
Vaccinum BCG
A vacina BCG liofilizada é uma vacina viva obtida a partir do cultivo do Bacilo de Calmette e Guérin, cepa atenuada de Mycobacterium bovis, de inocuidade e eficácia reconhecidas, para conferir proteção ao homem contra a tuberculose. O liofilizado é massa bacilar dessecada, com consistência de pó, de cor esbranquiçada ou amarelo pálido que, quando reconstituída, se apresenta ligeiramente turva e de aspecto homogêneo.
A produção da vacina é baseada no sistema de lote-semente, não podendo ser realizados mais do que oito subcultivos a partir da cepa original. A cepa selecionada deve conservar sua estabilidade e manter seu caráter não-patogênico tanto para o homem quanto para animais de experimentação. Essa vacina deve ser produzida por equipe em boas condições de saúde, que não trabalhe com agentes infecciosos e, em particular, com cepas virulentas de Mycobacterium tuberculosis.
A bactéria é inoculada em meio de cultura apropriado, isento de substâncias que possam causar reações tóxicas e/ou alérgicas ao ser humano. Os cultivos e o meio de cultura de cada recipiente são examinados visualmente quanto ao aspecto, apresentando véu bacteriano na superfície e meio de cultura límpido. Os cultivos são transferidos para novo meio e, após crescimento, são testados quantoà esterilidade e avaliados visualmente quanto à transparência do meio e aspecto do véu bacteriano. Após a filtração do véu bacteriano, esteé ressuspendido em meio apropriado e submetido aos testes de respiração bacteriana, opacidade e esterilidade. A suspensão bacteriana é diluída para o número apropriado de doses e, antes de proceder ao envase, o produto é avaliado quanto ao número de unidades formadoras de colônias e esterilidade. O produto é envasado em ampolas ou frascos-ampola de vidro âmbar classe farmacêutica, liofilizado, rotulado e submetido aos controles requeridos.
IDENTIFICAÇÃO
Observar por microscopia esfregaço obtido após a reconstituição da vacina e corado pela técnica de Ziehl-Nielsen. São detectados somente bacilos álcool-ácido resistentes. Como complemento, observar a morfologia das colônias semeadas no meio de Lowenstein-Jensen, utilizado no ensaio microbiológico (unidades formadoras de colônias).As colônias são rugosas, predominantemente espraiadas e nãopigmentadas.
CARACTERÍSTICAS
pH (V.2.19). 5,5 a 7,0. Determinar após a reconstituição da vacina com diluente apropriado.
Homogeneidade. Utilizar a lâmina preparada na Identificação para verificar a dispersão dos bacilos na suspensão da vacina por escala de valores que variam de zero a seis. No máximo grau cinco.
| Grau | Estado de Agregação |
| 0 | Somente bacilos dispersos |
| 1 | Predominância de bacilos dispersos; alguns grumos pequenos |
| 2 | Predominância de bacilos dispersos; alguns grumos pequenos e médios |
| 3 | Bacilos dispersos e grumos pequenos |
| 4 | Bacilos dispersos e grumos pequenos e médios |
| 5 | Bacilos dispersos e grumos pequenos, médios e grandes |
| 6 | Grumos médios e grandes |
ENSAIOS FÍSICO-QUÍMICOS
Umidade residual. Proceder conforme descrito na monografia de Vacinas para uso humano. No máximo 3%.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade. Proceder conforme descrito na monografia de Vacinas para uso humano.
Micobactérias virulentas. Reconstituir o conteúdo das ampolas com o diluente recomendado, de forma a se obter 50 doses humanas. Inocular volume de 1 ml em cada uma de seis cobaias, pesando de 250 g a 400 g, por via subcutânea, na região abdominal, do lado direito.
Manter os animais em observação por 42 dias. Ao final do período, pesar, sacrificar e necropsiar os animais. Examinar o local da inoculação, os gânglios regionais, inguinais, axilares, mediastínicos, lombares, portal e demais órgãos, em particular os pulmões, fígado, baço e rins.
Nenhuma cobaia pode apresentar evidência de tuberculose progressiva e, pelo menos, 2/3 dos animais têm que sobreviver ao final do período de observação, com ganho de peso. Repetir o teste de mais que 1/3 dos animais morrerem ou perderem peso.
Reatividade cutânea. Reconstituir uma amostra e preparar diluições 1:10 e 1:100, utilizando o diluente recomendado. Inocular, por via intradérmica, 0,1 ml de cada uma das diluições no flanco esquerdo de quatro cobaias albinas de mesmo sexo, com peso mínimo de 350 g cada. Os animais têm que apresentar reação tuberculínica negativa, bem como não podem ter sofrido tratamento que possa dar falso negativo. Proceder conforme descrito para a vacina de referência, inoculando o mesmo animal no flanco direito. Observar os animais por quatro semanas e realizar leituras semanais do diâmetro das lesões encontradas nos pontos de inoculação. Ao final do período de observação, calcular, para cada diluição correspondente, a média das quatro leituras da vacina e da vacina de referência. A vacina cumpre o requisito se a reação produzida pela amostra é semelhante à da vacina de referência.
ENSAIO MICROBIOLÓGICO
Número de unidades formadoras de colônias (UFC). Reconstituir cinco ampolas da vacina com diluente recomendado, tendo o cuidado de adicioná-lo suavemente para evitar a formação de espuma. Transferir o conteúdo das ampolas para um único tubo de ensaio, homogeneizar e proceder três diluições, de modo a obter número ótimo de colônias em torno de 40, desprezando as contagens superiores a 100. Inocular em meio Lowenstein-Jensen, utilizando cinco tubos para cada uma das duas diluições mais concentradas e 10 tubos para a mais diluída. Vedar os tubos e incubar na posição vertical à temperatura de 37 ºC, por quatro semanas. Analisar em paralelo uma amostra da vacina de referência. Os limites são 2 x 106 a 10 x 106 UFC/ml.
TERMOESTABILIDADE
Incubar cinco ampolas da vacina à temperatura de 37 ºC por quatro semanas e proceder ao Ensaio microbiológico. Comparar os resultados obtidos com os das amostras mantidas à temperatura de 2 ºC a 8 ºC. O número de UFC/ml não pode ser inferior a 20% de UFC/ml da vacina mantida entre 2 ºC e 8 ºC.
EMBALAGEM E ARMAZENAMENTO
Cumpre o estabelecido na monografia de Vacinas para uso humano.
ROTULAGEM
Observar a legislação vigente.
124
VACINA DE VÍRUS VIVOS CONTRA FEBRE AMARELA
Vaccinum febris flavae vivum
A vacina contra febre amarela é constituída de vírus vivos atenuados
e apresentada sob a forma liofilizada. Após a reconstituição com
diluente apropriado, tem aspecto de suspensão homogênea, podendo
demonstrar coloração devido à presença de indicador de pH.
A produção da vacina é baseada no sistema de lote-semente primário
da estirpe 17D, do qual, por meio de passagens em ovos embrionados
de galinha SPF (livre de microrganismos contaminantes), se origina o
lote-semente secundário. Esse lote deve ser avaliado quanto à neurovirulência
em macacos suscetíveis e não pode apresentar microrganismos
estranhos. Além disso, a cepa viral tem que demonstrar
imunogenicidade adequada e segurança para o ser humano.
A replicação do vírus é realizada em ovos embrionados de galinha, livre de patógenos específicos, ou em cultura de células suscetíveis. A suspensão viral é identificada e controlada quanto à esterilidade. Após a clarificação da suspensão viral por método adequado para remoção de resíduos celulares, algumas substâncias estabilizadoras, que, comprovadamente, não alteram a eficácia e segurança do produto, são adicionadas. Antes do envase e liofilização, o produto é analisado quanto à esterilidade, concentração de vírus e nitrogênio protéico. Se a produção da vacina ocorrer em ovos embrionados, 2% e não menos que 20 ovos são separados para controle. Ao final da produção da vacina, estes ovos não-infectados com a cepa vacinal têm que demonstrar ausência de patógenos específicos para aves.
O produto é envasado em recipientes adequados, liofilizado, rotulado e submetido aos controles requeridos.
Outras informações relativas aos critérios de produção e seus controles estão indicadas na monografia de Vacinas para uso humano.
IDENTIFICAÇÃO
A vacina, quando neutralizada com soro contendo anticorpo específico para o vírus da febre amarela, inibe a formação de unidades formadoras de “plaque” UFP) em células suscetíveis conforme descrito em Determinação de potência.
ENSAIOS FÍSICO-QUÍMICO
Umidade residual. Proceder conforme descrito na monografia de Vacinas para uso humano. No máximo 3%.
TESTES DE SEGURANÇA BIOLÓGICA
Esterilidade. Proceder conforme descrito na monografia de Vacinas para uso humano.
Endotoxina bacteriana (V.5.1.9). Cumpre o teste. No máximo 10 UE/ml.
Ovalbumina residual. Três frascos de um mesmo lote de vacina liofilizada
e ovalbumina padrão são submetidos ao método imunoenzimático
ELISA. A curva padrão de ovalbumina é feita nas concentrações
de 100 a 0,5 μg com diluições usando fator 2 e a vacinaé diluida em PBS/T20 0,05%/NFDM (salina tampão fosfato com
tween 20 e leite em pó desnatado). Inocular cada diluição iniciando
em 1:10 e usar o fator 2 em dois orifícios da placa de 96 orifícios
previamente sensibilizada com soro anti-ovalbumina (coelho) em
tampão carbonato-bicarbonato de sódio e bloqueada com soro albumina
bovina 3%. A incubação é feita por 30 minutos a 37 ºC. As
placas são lavadas com PBS/T20 0,05% (salina tampão fosfato com
tween 20) e o soro anti-ovalbumina (coelho) conjugado a peroxidase
em PBS/T20 0,05%/NFDM é adicionado. É feita nova incubação por
30 minutos a 37 ºC e nova lavagem. A reação é revelada com o
substrato para a peroxidase em tampão citrato-fosfato e é paralisada
com ácido sulfúrico 2 M. A leitura é feita em leitor de microplacas a
um comprimento de onda de 450/630 nm.
O teor de ovalbumina residual é calculado plotando a média da
absorvância contra o log da concentração padrão usando valores lineares
correspondentes a 50% do “endpoint”.
A vacina é considerada satisfatória se o conteúdo de ovalbumina residual for menor ou igual que 5 μg/dose.
DETERMINAÇÃO DA POTÊNCIA
Pelo menos dois frascos de vacina liofilizada e um de vacina referência são submetidos ao método de unidades formadoras de “plaque” (UFP). Diluir a vacina aplicando-se fator 4 e inocular cada diluição em, pelo menos, três orifícios em placa de seis orifícios contendo monocamada de células Vero previamente semeadas. A concentração da linhagem celular pode variar de 150 000 a 300 000 células por ml, conforme o dia de sua utilização. Após adsorção por período de 90 minutos à temperatura de 36 ºC, em ambiente de CO2 a 5%. Os inóculos são retirados e um meio de cultura, contendo agarose ou carboximetilcelulose em concentração adequada, é adicionado.
As células são incubadas por cinco a sete dias, à temperatura de 36 ºC, em ambiente de CO2 a 5%. Após o período de incubação, retirar o meio de crescimento, fixar as células com formaldeído e corar com um corante vital.
A potência da vacina é calculada pela média do número de plaques de, pelo menos, duas diluições e o resultado, expresso em log10 UFP/dose. Para a determinação ser considerada válida, é necessário que (a) o controle de cultura de células apresente monocamada inalterada;(b) a variação de potência entre as duas amostras da vacina não seja maior que 0,5 log10 UFP; (c) a potência da vacina de referência não varie mais que 0,5 log10 UFP do seu título médio; (d) o número de UFP seja decrescente em relação às diluições crescentes.
A potência em UFP/dose tem que ser equivalente a 1 000 DL50 em camundongos. Caso a amostra não cumpra com os requisitos, repetir a determinação. A potência da vacina é a média geométrica das duas determinações realizadas.
TERMOESTABILIDADE
O teste é realizado em paralelo à Determinação de potência. Incubar, pelo menos, dois frascos de vacina por 14 dias a 36 ºC e analisar conforme descrito em Determinação de potência. A vacina não pode perder mais que 1 log10 UFP em relação ao título determinado na amostra conservada em condições adequadas de temperatura. Além disso, não apresentar título inferior ao especificado para a potência do produto.
EMBALAGEM E ARMAZENAMENTO
Cumpre o estabelecido na monografia de Vacinas para uso humano.
ROTULAGEM
Observar a legislação vigente.
________________________________________________
XII.2 REAGENTES E SOLUÇÕES REAGENTES
Soro anti-ovalbumina (coelho)
Preparação- Imunizar coelhos com ovalbumina numa concentração de 20 μg/dose com adjuvante completo de Freud em três doses com um intervalo de 15 dias cada dose. Sangrar os coelhos após 7 dias e testar o soro em um ELISA com um conjugado anti-ovalbumina preparado em uma diluição 1:10.
Soro anti-ovalbumina (coelho) conjugado a peroxidase Preparação- Utilizar o soro anti-ovalbumina (coelho) e conjugá-lo a peroxidase. E testar em um ELISA para validá-lo.
Soro Albumina Bovina - BSA 3%
Preparação- Dissolver 15 gramas de Soro albumina bovina (BSA) em 500 ml de água destilada. Filtrar em filtro 0,22 μm. Distribuir em frascos com 40 ml cada. Estocar a - 20ºC.
Ovalbumina Padrão (100 μg/ml)
Preparação- Dissolver 1 g de ovalbumina Grau VI em 10 ml de água destilada (100 mg/ml). Misturar bem sem agitação para evitar a formação de espuma. Diluir 100 μl da albumina 100 mg/ml em 100 ml de água para obter uma concentração de 100 μg/ml. Armazenar em alíquotas e conservar a -20 ºC.
Àcido Sulfúrico 2 M
Preparação- Adicionar 55 ml de ácido sulfúrico em 500 ml de água destilada. Estocar a temperatura ambiente.
XII.4 TAMPÕES
Tampão carbonato-bicarbonato de sódio pH 9.6
Preparação- Dissolver 0,75 g de carbonato de sódio e 1,5 g de bicarbonato de sódio em 500 ml de água. Distribuir em frascos com 50 ml cada. Autoclavar a 121 ºC - 1 atm por 20 minutos. Estocar a 4ºC.
Tampão citrato-fosfato pH 5.0
Preparação- Misturar com agitação as soluções A e B até ajustar o pH para 5,0. Distribuir em frascos com 50 ml cada. Autoclavar a 121 ºC - 1 atm por 20 minutos. Estocar a 4 ºC.
Solução A: dissolver 0,8 g de fosfato de sódio dibásico heptahidratado em 500 ml de água.
Solução B: dissolver 3,5 g de ácido cítrico monoidratado em 500 ml de água
Distribuir em frascos com 50 ml cada. Autoclavar a 121 ºC, - 1 atm por 20 minutos. Estocar a 4 ºC.
PBS - Salina tampão fosfato
Preparação- Dissolver com agitação 24 g de cloreto de sódio 0,6 g de cloreto de potássio, 4,3 g de fosfato de sódio dibásico dodecaidratado e 0,6 g de fosfato monobásico de potássio em 4 litros de água.
Autoclavar a 121 ºC - 1 atm por 20 minutos. Estocar a 4 ºC.